1) Korozja – podstawy
Korozja to chemiczne i elektrochemiczne procesy stopniowego niszczenia mikrostruktury materiału prowadzące do jego rozpadu pod wpływem czynników środowiska. Określenie to związane jest zwłaszcza z metalami (stal), ale korozji ulegają zasadniczo wszystkie tworzywa: beton, polimerowe itp. Straty z powodu korozji są ogromne, dlatego bardzo ważne jest opracowanie skutecznych metod zapobiegania jej.
Ważnym materiałem konstrukcyjnym jest stal, która ze względu na właściwości fizyczne jest doskonałym materiałem do tego celu, gdyż stal nie zmienia swoich właściwości użytkowych w czasie (pomijając zmęczenie materiału oczywiście). Niestety żelazo, jak każdy metal o potencjale niższym niż wodór, reaguje z wodą dając wodorotlenek. Żelazo łatwo też reaguje z tlenem atmosferycznym. Procesy te, czyli korozja, skutkują nadżeraniem powierzchni żelaza i pokrywania się rdzą – produktem korozji. Wpływa to na wytrzymałość stali, gdyż ilość stali podczas korozji maleje. Korozja elektrochemiczna związana jest z utworzeniem się na powierzchni metalu ogniwa korozyjnego. Jedno miejsce stanowi strefę anodową, sąsiednie katodową. W takiej sytuacji korozja rozpoczyna się samorzutnie. Stal może absorbować na swoje powierzchni warstewkę wody, zwłaszcza w temperaturze poniżej punktu rosy na jej powierzchni, jako że jest hydrofilowa, kondensuje para wodna. Woda ta rozpuszcza w sobie elektrolity, obecne na powierzchni stali jako ślady, czy w powietrzu (gazy kwaśne, koloidalne cząstki soli itp.). Elektrolit zwiększa przewodnictwo wody a to sprzyja szybkości korozji.
Mechanizm korozji beztlenowej:
Anoda: Fe Fe2+ + 2e–
Katoda: 2H+ + 2e– H2
Sumarycznie: Fe+2H2O Fe(OH)2 + H2
Jednak ze względu na polaryzację układu w strefie katody (niedomiar elektronów) i wymagany przepływ elektronów z anody do katody taka korozja zatrzymuje się, chyba że stężenie kationów wodorowych jest duże (niskie pH). Jednakże w obecności tlenu następuje depolaryzacja katody, co znaczy, że ładunek elektryczny katody równoważy ładunek elektryczny anody:
Anoda: Fe Fe2+ + 2e–
Katoda: O2 + H2O+4e- 4OH–
Sumarycznie: 2Fe + O2 + 2H2O 2Fe2+ + 4OH– 2Fe(OH)2
Obecność miejsc o charakterze katodowym i anodowym spowodowana jest niejednorodnością potencjału elektrochemicznego powierzchni stali. Miejsca o wyższym potencjale mają charakter katodowy, miejsca o potencjale niższym – anodowy. Czynnikiem wpływającym na wartość potencjału są: lokalnie występujące różnice w naprężeniach wewnętrznych materiału, lokalne różnice składu materiału, lokalne wytrącenia, wtrącenia zanieczyszczeń na powierzchni metalu oraz stopień pokrycia powierzchni przez pasywną warstwę tlenku żelaza (III). Charakter poszczególnych miejsc na powierzchni można określić za pomocą odpowiednich pomiarów lub wskaźnika ferrochromowego, który w miejscach katodowych zabarwia się na różowo, a w anodowych na niebiesko.
Czynniki wpływające na szybkość korozji:
– Rodzaj materiału: poszczególne rodzaje stali korodują z różną szybkością, zależnie od ich składu. Niektóre są praktycznie odporne na korozję, zawierają dodatki stopowe podnoszące odporność, np. w stalach nierdzewnych, odpornych na korozję atmosferyczną, czy stale kwasoodporne które są odporne na trawienie przez silne kwasy dzięki strukturze austenitycznej. Jednakże stale te mają tylko specjalne zastosowania i ze względu na cenę nie są używane jako podstawowy materiał konstrukcyjny.
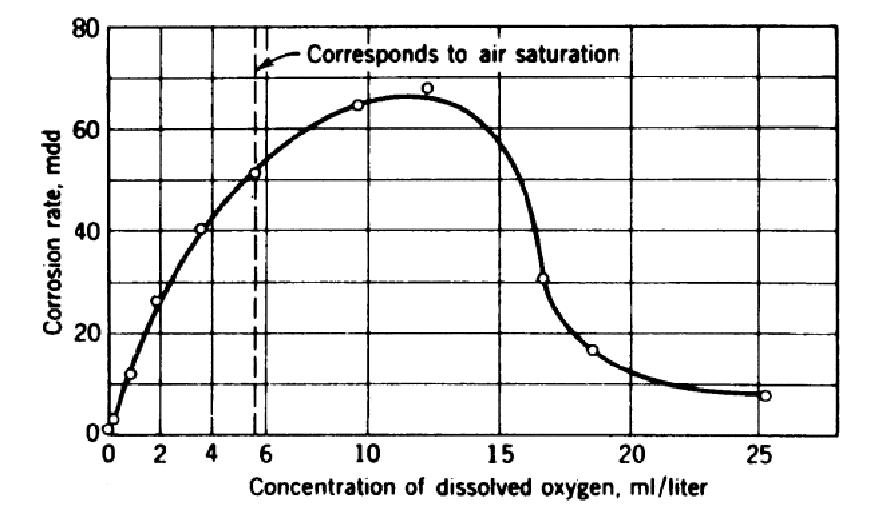
– Stężenie tlenu: szybkość korozji zależy od stężenia tlenu rozpuszczonego w wodzie i docierającego do powierzchni stali. Pokazuje to poniższy rysunek
W niższych stężeniach szybkość korozji rośnie, ale osiąga maksimum i przy jeszcze wyższych spada, z powodu pasywacji. Linia przerywana odpowiada rozpuszczalności tlenu atmosferycznego w wodzie w 25 oC i 1013 hPa. [1]
– Przewodnictwo elektrolitu na powierzchni stali: szybkość zależy od stężenia elektrolitu. Tak jak i w poprzednim przypadku i tutaj szybkość korozji przechodzi przez maksimum, jak na rysunku.

Rysunek pokazuje szybkość korozji w nasyconej tlenem atmosferycznym solance, zależnie od stężenia soli. Spadek szybkości korozji poza maksimum związany jest z gwałtownym spadkiem rozpuszczalności tlenu przy większych stężeniach soli. Początkowo jednak wzrost stężenia solanki powoduje przyśpieszenie korozji. Linia przerywana oznacza średnie stężenie soli w wodzie morskiej i jak widać, jest to także stężenie, które w największym stopniu sprzyja korozji [1]. Ponieważ elektrolit, którym może pokryć się stal podczas kondensacji pary wodnej zawiera małe ilości składników jonowych, przeto ich obecność sprzyja korozji.
– Wpływ kwasowości środowiska (pH): szybkość korozji zależy od stężenia jonów wodorowych w elektrolicie. Żelazo wypiera wodór z kwasów nieutleniających, zatem ze wzrostem ich stężenia (kwasowości) rośnie szybkość roztwarzania żelaza. Jednakże przy pH = 4 i więcej szybkość korozji spada i aż do pH = 10 praktycznie nie zależy od pH. W tym obszarze pH wytrąca się Fe(OH)2 i pokrywa powierzchnię stali.

W tym przedziale pH szybkość korozji jest określona przez szybkość dyfuzji tlenu przez warstwę wodorotlenku do stali. Stal styka się pod warstwą wodorotlenku w r-rem o pH ok. 9,5. Wzrost zasadowości powyżej pH = 10 podnosi także pH przy powierzchni stali. Szybkość korozji spada, gdyż zaczyna się proces pasywacji stali [1]. Natomiast poniżej przedstawiono wykres E-pH (diagram Pourbaix) dla żelaza. Diagram odnosi się do metalu zanurzonego w czystej wodzie a jedynymi stałymi składnikami są tlenki żelaza i żelazo. W rzeczywistości zakres przedziałów pH korozji/pasywacji jest funkcją stężenia jonów metalu (Fe2+).
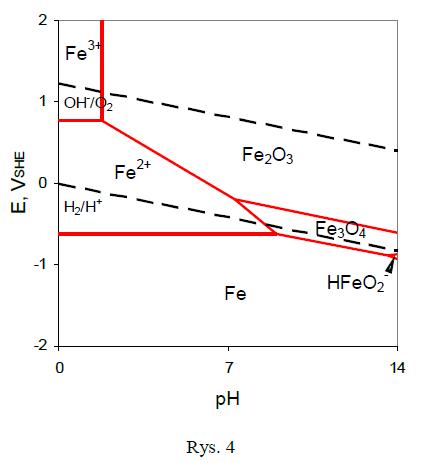
Pomiędzy liniami przerywanymi znajduje się obszar termodynamicznej trwałości wody. Przy niskich potencjałach w całym zakresie pH (w dolnej części wykresu) żelazo jest trwałe w formie metalicznej (jako atom Fe). To oznacza brak reakcji utleniania tego metalu. To jest obszar odporności żelaza. W tych warunkach żelazo nie ulega korozji elektrochemicznej. Przy wyższych potencjałach w roztworach kwaśnych (o niskim pH) trwałymi formami żelaza są jego jony Fe2+ i Fe3+. W tych warunkach żelazo będzie ulegać reakcjom korozyjnym, takim jak reakcja (1). To jest obszar korozji żelaza. W prawej, górnej części wykresu (wysokie potencjały, roztwory obojętne i zasadowe) jest szeroki obszar trwałości tlenków żelaza. W tych warunkach żelazo będzie się utleniać do tlenków, na przykład wg reakcji (2). Powstające produkty korozji (tlenki) mogą utworzyć warstwę pasywną na powierzchni metalu. To jest obszar pasywności żelaza, a więc znikomej szybkości jego korozji. Przy pH zbliżonych do 14 korozja może nastąpić ze względu na roztwarzanie
Interpretacja wykresów Pourbaix polega na określeniu warunków w jakich dany metal jest odporny na korozję, w jakich ulega jonizacji a w jakich staje się pasywny. Każdy metal posiada potencjał jonizacji, np. dla żelaza jest to potencjał reakcji Fe/Fe2+ oraz potencjał utlenienia do tlenku Fe/Fe2O3. Wartości obu tych potencjałów zależą od pH. Jednakże potencjał jonizacji słabo zależy od pH, potencjał utlenienia silnie. W związku z tym proste wyznaczające wartości odpowiednich potencjałów od pH mogą się przeciąć (punk A na rys. 5). Przy pH powyżej punktu A łatwiej jest metal utlenić niż zjonizować. Obszar C oznacza roztwarzanie (jonizację) metalu, czyli jego korozję, obszar I oznacza odporność na korozję natomiast obszar P to pasywność metalu i odporność na korozję także.

Natomiast kolejny rysunek ma dodatkowo naniesiony obszar trwałości wody. Obszar dodatkowo podzielono na C1 i C2. C1 to obszar, w którym korozja zachodzi poprzez redukcję tlenu, C2 to obszar w którym korozja zachodzi na zasadzie wyparcia wodoru. Również i w tym przypadku proste się przecinają. Ponad punktem przecięcia się potencjału wydzielania wodoru potencjał jonizacji metalu jest wyższy niż potencjał wodoru i metal traci możliwość reakcji z wodą, nie traci natomiast możliwości reakcji z tlenem. Od tego punktu zachodzi tylko korozja na zasadzie reakcji z tlenem.

Zatem jeśli uwzględnimy obszary C1 i C2 na wykresie dla żelaza to rysunek 4 przyjmie postać jak na rysunku 7. Wyróżniono na nim typy korozji w zależności od warunków.
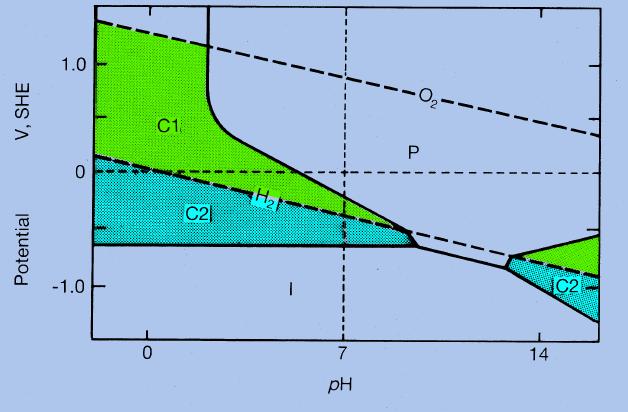
Rysunek ten skomplikowałby się jeszcze bardziej gdyby narysować na nim przebieg lini dla kilku różnych stężeń, ale dotyczy to głównie zakresu pola roztwarzania żelaza. Wykresy Pourbaix dla innych metali zostaną przedstawione w rozdziale poświęconym ochronie barierowej.
Na przebieg zakresów obszarów C, I, P wpływa temperatura, gdyż potencjał jest funkcją temperatury (równanie Nernsta). Poniżej przedstawiono wykresy Pourbaix dla Fe w 25 i 200 oC.

Jak widać, obszar pasywności jest węższy, kwasowe właściwości tlenków żelaza wzrosły.
UWAGA: wykresy Pourbaix nie mówią o szybkości korozji, pozwalają jedynie określić warunki w których zachodzi. Nie dają 100% pewności, pozwalają jedynie prognozować.
Wygodnym sposobem określania szybkości reakcji elektrochemicznych jest pomiar natężenia prądu tych reakcji. Zgodnie z pierwszym prawem Faraday’a ubytek masy metalu (ubytek korozyjny) jest proporcjonalny do natężenia prądu reakcji, której ten metal ulega, na przykład reakcji (1) lub (2). Aby wyznaczyć szybkość reakcji korozyjnych metalu w danym roztworze korozyjnym (o określonym pH) mierzona jest zależność natężenia prądu reakcji lub gęstości prądu reakcji (natężenie prądu podzielone przez pole powierzchni metalu, na którym zachodzi reakcja) od potencjału metalu. Ta zależność, zwana krzywą polaryzacji, jest rejestrowana przy wymuszonej zmianie potencjału metalu (polaryzacji) w roztworze korozyjnym. Krzywa polaryzacji otrzymana przy zwiększaniu potencjału metalu (badane są wtedy jego reakcje korozyjne) jest nazywana krzywą polaryzacji anodowej.
Analiza przebiegu krzywych polaryzacji anodowej pozwala określić, jakim reakcjom korozyjnym ulega metal w badanym roztworze w różnych zakresach potencjału. Na rys. 3. przedstawiono schematycznie typowe przebiegi krzywych polaryzacji. Metal wykazuje w roztworze charakterystyczny potencjał Eo, przy którym prąd nie płynie (w rzeczywistości przy tym potencjale prąd reakcji utleniania jest równy prądowi reakcji redukcji). Przy potencjałach niższych od Eo reakcje utleniania metalu zachodzą bardzo wolno i można przyjąć, że jest to potencjałowy zakres odporności metalu. Reakcje anodowe dominują przy potencjałach wyższych od Eo. Szybki wzrost prądu przy zwiększaniu potencjału (rys. 3a) odpowiada reakcji korozyjnej, w której metal utlenia się do jonu metalu – reakcja (1). Jest to więc potencjałowy zakres korozji metalu. Natomiast nieznaczne natężenie prądu, mało zależne od potencjału (rys. 3b) odpowiada utlenianiu metalu do tlenku, na przykład wg reakcji (2). Ta niewielka gęstość prądu wynika ze znikomej szybkości reakcji korozyjnej zachodzącej na metalu pokrytym warstwą pasywną. Taki przebieg krzywej polaryzacji wyznacza potencjałowy zakres pasywności metalu. Na rysunkach a), b), c) przedstawiono różne typy przebiegu krzywych polaryzacji.


Rysunek c) dotyczy żelaza. Występujące maksimum nazywamy krytycznym potencjałem pasywacji, to jest potencjał, przy którym powierzchnia metalu zaczyna pokrywać się warstwą pasywną. Gęstość prądu spada i ustala się na pewnej wartości, staje się niezależna od przyłożonego potencjału. ten punkt przegięcie nazywamy normalnym potencjałem pasywacji i oznacza on punkt, w którym powierzchnia metalu jest całkowicie pokryta warstwą pasywną.
Jak wyjaśniono, pasywacja żelaza zachodzi przy pH = 10 i więcej. Nasycony r-r Ca(OH)2 wykazuje pH ok. 11,2 stąd stalowe zbrojenia w betonie wykazują znaczną odporność na korozję, póki pH nie spadnie poniżej krytycznej wartości. Ca(OH)2 ze względu na wielokrotnie większą rozpuszczalność niż inne składniki betonu jest praktycznie jedynym związkiem wpływającym na pH zaprawy. Niestety jest też systematycznie wypłukiwany przez wodę, co z czasem skutkuje spadkiem pH i korozją prętów zbrojeniowych, zwiększeniem ich objętości spowodowanej produktami korozji i ostatecznie popękaniem konstrukcji betonowej.
UWAGA: Wykresy Pourbaix mają inny przebieg w obecności innych elektrolitów, np. NaCl, jest możliwa korozja stali w pH ponad 10. Aniony kwasów nieorganicznych utrudniają tworzenie się warstwy pasywnej, a chlorkowe nawet ją niszczą. Stąd jony chlorkowe należą do bardzo agresywnych czynników (korozja chlorkowa). W obecności chlorków intensywnie koroduje aluminium. W wodzie morskiej aluminium ulega szybszej korozji niż stal. Aluminium pokryte jest bardzo szczelną powłoką i praktycznie nie ulega korozji atmosferycznej, choć jest bardzo reaktywny chemicznie. Warstewka ta chroni metal w obojętnym środowisku do tego stopnia, że glin praktycznie nie reaguje z r-rem CuSO4. Dodatek NaCl wielokrotnie przyspiesza tę reakcję.
– Wpływ temperatury: jak wiadomo, wzrost temperatury zwiększa szybkość reakcji. Nie inaczej jest w przypadku korozji. Szybkość korozji zależy jednak od tego, czy korozja zachodzi w układzie otwartym czy zamkniętym. Z jednej strony temperatura powoduje wzrost szybkości reakcji, z drugiej jednak rozpuszczalność tlenu w wodzie silnie maleje ze wzrostem temperatury, co ogranicza jej szybkość. W efekcie jeśli jeśli próby przeprowadzić w układzie otwartym, co umożliwia swobodnie opuszczenie wody przez tlen (p=const.), pojawi się maksimum na krzywej korozji. W układzie zamkniętym, podczas ogrzewania stężenie tlenu, którego ciśnienie w układzie rośnie, jest ciągle duże i szybkość korozji jest liniową funkcją temperatury [1].

Czynniki agresywne w wodzie:
Składnik lub charakterystyka wody | Wpływ na szybkość korozji |
Odczyn pH Rozpuszczone gazy: -tlen – dwutlenek węgla Rozpuszczone sole: -jony agresywne -jony metali -jony nieagresywne – całkowita ilość substancji rozpuszczonej | Dla większości wód naturalnych odczyn pH nie ma wpływu na szybkość korozji. W zakresie pH 4,5 -9,0 szybkość korozji jest prawie stała. Tlen jest efektywnym depolaryzatorem katodowym i w związku z tym szybkość korozji wzrasta wraz ze wzrostem jego stężenia (typowe zawartości 2-10 mg/1). Przy wysokich stężeniach może wystąpić pasywujące działanie tlenu, chociaż działanie to uniemożliwiają rozpuszczone sole, a w szczególności jony chlorkowe. Działanie tlenu jest uzależnione od wielu innych czynników, a mianowicie: temperatury, przepływu i składu chemicznego wody. W przypadku nierównomiernego wprowadzenia tlenu mogą się tworzyć ogniwa stężeniowe. Dwutlenek węgla może wpływać bezpośrednio (co wynika z jego kwaśnego charakteru), stanowiąc źródło jonów wodorowych dla reakcji katodowej i pośrednio ograniczając tworzenie się węglanu wapniowego. Jony siarczanowe i chlorkowe są zawsze agresywne, gdyż utrudniają tworzenie się warstw ochronnych. Jony chlorkowe mogą niszczyć stan pasywny, co prowadzi do korozji wżerowej. Jony niektórych metali (np. miedzi) mogą powodować tworzenie lokalnych ogniw bimetalicznych wskutek redukcji i wydzielania się na powierzchniach stalowych i tym samym mogą przyspieszać korozję lokalną. Jony takie jak wapń, magnez, kwaśne węglany – mają własności inhibitujące. Wielkość zasolenia wody ma wpływ głównie na szybkość korozji układów bimetalicznych. Jest potrzebna przy badaniu przydatności ochrony katodowej. |
Jednak niektóre elektrolity ze względu na utleniające właściwości (HNO3, CrO3) czy też niską rozpuszczalność i wytrącanie się na powierzchni metalu ograniczają szybkość korozji. Utlenicze mogą powodować pasywację, zaś np. wodorowęglany Ca i Mg w wodzie twardej mogą wytrącać się jako CaCO3 i MgCO3 na powierzchni metalu ograniczając kontakt środowiska z metalem.
2) Ochrona przed korozją
Istnieją dwie zasadnicze metody ochrony przed korozją:
– anodowa: czyli zatrzymanie reakcji anodowej
– katodowa: sam metal staje się katodą i jest redukowany przez elektrony z anody.
Ochrona katodowa
Jeśli połączyć stal do dodatniego bieguna baterii to sama staje się katodą. Jeśli obie elektrody zanurzyć w elektrolicie, to stal nie koroduje ze względu na nadmiar potencjału. Skutkiem jest elektroliza wody i na katodzie wydziela się H2. Metody ochrony katodowej polegają na połączeniu stali z metalem o niższym potencjale (aluminium, cynkiem, magnezem). Stal pokryta warstwą cynku jest dobrze zabezpieczona przed korozją, nawet jeśli powłoka cynkowa jest porowata, gdyż na stali gołej powierzchni stali wydziela się wodór, a więc stal jest chroniona przed utlenianiem. Cynk można nanosić metodami galwanicznymi lub ogniowymi. Aluminium, choć znacznie aktywniejsze niż cynk, nie jest zbyt skuteczne, ze względu na swoją pasywację. Warstwa pasywna na tyle podnosi potencjał glinu, że stal staje się wrażliwa na korozję. Powłoka z aluminium musiałaby być szczelna. Aluminium dobrze sprawdza się natomiast w wodzie morskiej, gdyż jak już wspomniano, chlorki niszczą warstwę pasywną glinu. Magnez ze względu na łatwość z którą ulega utlenianiu dobrze sprawdza się w roli protektorów konstrukcji stalowych, podobnie jak i cynk. Protektory nie są jednak w stanie chronić konstrukcji zbyt dużych a samo zaplanowanie ich rozmieszczenia może stanowić problem techniczny.
Cynk pokrywa się warstewką tlenku i węglanu (zasadowego węglanu) na powietrzu i wymaga mniej niż 6 ml/dm3 tlenu w wodzie do pasywacji. Jednakże związki te wykazują rozpuszczalność w wodzie i warstewka ta jest systematycznie wypłukiwana przez wodę, co powoduje, że cynk koroduje, ale wolniej niż niechroniona stal i dlatego jest w stanie chronić stal przez pewien okres czasu, zależnie od środowiska korozyjnego. Tlenek cynku ze względu na amfoteryczny charakter nie jest odpory ani na kwasy, ani na alkalia, więc kwaśne deszcze znacznie przyśpieszają jego korozję.
Ochrona anodowa: pasywacja
Jak pokazano na rysunku 1, wzrastające stężenie tlenu powoduje do ok. 12 ml/dm3 zwiększa szybkość korozji przez depolaryzację katody. Jednakże przy wyższych stężeniach więcej tlenu dosięga powierzchni metalu niż się redukuje w reakcji katodowej i korozja zostaje wstrzymana. Mechanizm wstrzymywania nie jest do końca wyjaśniony, ale teoria zakłada, że nadmiar tlenu powoduje natychmiastowe utlenienie jonów Fe2+ z reakcji katodowej do Fe3+. W połączeniu z OH– tworzy się Fe(OH)3, który jest znacznie mniej rozpuszczalny w wodzie niż Fe(OH)2 i bariera z uwodnionego Fe(OH)3 tworzy się na powierzchni metalu. Zatrzymanie korozji przez wstrzymanie reakcji anodowej nazywane jest pasywacją metalu.
Krytyczne stężenie tlenu dla pasywacji zależy od warunków. Rośnie ono ze stężeniem rozpuszczonych soli (poprzedni rozdział, korozja chlorkowa) i z temperaturą. Natomiast maleje ze wzrostem pH i z szybkością przepływu wody nad powierzchnią metalu. Przy pH ok. 10 sięga ono 6 ml/dm3 co odpowiada stężeniu nasyconej powietrzem wody i ze wzrostem pH jeszcze spada. Żelazo jest spasywowane przeciwko korozji przez tlen przy pH powyżej 10. Kontrolowanie korozji żelaza w niższych wartościach pH jest niepraktyczne, gdyż stężenia krytyczne stają się większe niż równowagowe stężenie tlenu z powietrza. Jednakże zamiast tlenu można użyć całą gamę innych związków jako pasywatory: chromiany, azotyny, molibdeniany, ołowiany wolframiany są tego przykładami. Podobnie jak w przypadku tlenu wymagane jest minimalne (krytyczne) stężenie tych czynników, mniejsze stężenia mogą powodować korozję przez depolimeryzację katody. Reakcje z chromianami są najlepiej poznane. Na powierzchni stali powstaje warstwa częściowo uwodnionych tlenków mieszanych chromu i żelaza trójwartościowego, która stanowi skuteczną barierę.
Pewne sole nieutleniające, np. sole metali alkalicznych kwasów borowego, fosforowego, węglowego, benzoesowego także są czynnikami pasywującymi. Ich działanie polega na ich zasadowym charakterze: obniżają krytyczne stężenie tlenu przez zwiększanie pH.
Całkiem nowym rozwiązaniem jest pasywacja z użyciem powłoki elektroprzewodzącego polimeru na stali . Polifenylenoamina, zwana polianiliną, jest dostępna pod handlowymi nazwami Zypan, Versicon, Panda, jest skuteczna gdyż prowadzi do otrzymania szczelnej, bardzo cienkiej tlenkowej warstwy pasywującej na powierzchni metalu. Proszki polimeru są nierozpuszczalne we wszelkich rozpuszczalnikach, nietopliwe i trudne do zdyspergowania z powodu bardzo wysokiego napięcia powierzchniowego. Elektropolimeryzacja i osadzanie polianiliny z buforu fosforanowego na powierzchni stali podnosi odporność na korozję stali. Stal pokryta politiofenem przez elektropolimeryzację także wykazuje ochronę przed korozją. Elektroprzewodząca powłoka z poli[2,5-bis(N-metylo-N-propylamino)fenylenowinylenu osadzona na aluminium także wzmaga jego odporność na korozję [3, 4].
Ochrona barierowa i inhibicja
Polega na pokryciu stali powłoką która skutecznie izoluje stal od środowiska (H2O i O2). Przykładem może być wspomniany cynk. Ale są także inne metale, które wykazują znaczną odporność na korozję i mogą służyć jako bariery. Przykładami są cyna, nikiel, miedź, tytan i chrom, ale także złoto, srebro, platyna. Trójka ostatnich ze względu na cenę i wyjątkowe walory dekoracyjne służy tylko dekoracji, lub do celów specjalnych, ale nie do zabezpieczania typowych konstrukcji. Oprócz czystych metali, można nakładać powłoki stopów, np. mosiądzu na stal. Miedź, srebro, złoto i platyna jako metale o potencjale wyższym niż wodór nie reagują z wodą i kwasami nieutleniającymi, ale dwa pierwsze z nich w obecności tlenu pokrywają warstewką tlenków, a w obecności dodatkowo wilgoci i CO2 miedź pokrywa się zielonkawym nalotem zasadowych węglanów zwanych ogólnie patyną. Na miedzi, jako na metalu o zmiennej wartościowości tworzy się układ: Cu – Cu2O – CuO. Zewnętrzną warstwę stanowi czarny tlenek miedzi (II).
Metale te dobrze zabezpieczają stal przed korozją, ale pod warunkiem szczelności powłoki. gdyż żelazo w kontakcie z metalem o wyższym potencjale szybko koroduje, jeśli ma kontakt z wodą i tlenem i innymi czynnikami korozyjnymi. Miedź stanowi lepszą barierę niż cynk, pod warunkiem szczelności. Dobrze chroni metal w środowisku obojętnym, koroduje powoli, odporna jest na kwasy nieutleniające, ale w obecności tlenu staje się na nie nieodporna, ze względu na utlenianie. Wykazuje znaczną odporność na alkalia, gdyż tlenki miedzi mają słabo amfoteryczny charakter, ale nie jest odporna na środowisko amoniaku i soli amonowych. W obecności tlenu i amoniaku miedź szybko się roztwarza przez tworzenie się rozpuszczalnego w wodzie [Cu(NH3)4]2+.
Wykresy E-pH dla metali stanowiących ochronę barierową stali:
Miedź:

Wykres pokazuje, że w wodzie pozbawionej tlenu miedź nie ulega korozji. Przy dużych stężeniach rośnie jej odporność na zasadowe środowisko, a także kwaśne. W roztworach zasadowych miedź koroduje w obecności tlenu jeśli są małe stężenia jonów CuO2- w r-rze.
Tytan:

Tytan wykuzuje bardzo dużą odporność na korozję, warstwę pasywną stanowi TiO2. Nie jest w pełni odporny na kwaśne środowisko. W wodzie nie zawierającej tlenu ulega korozji redukując wodór, w obecności tlenu jednak ulega całkowitej pasywacji.
Glin:

Wykres potwierdza że w środowisku obojętnym metal ten jest bardzo odporny na korozję ze względu na silną pasywację. Jednak w środowisku kwaśnym lub zasadowym chętnie koroduje.
Chrom (symulacja):
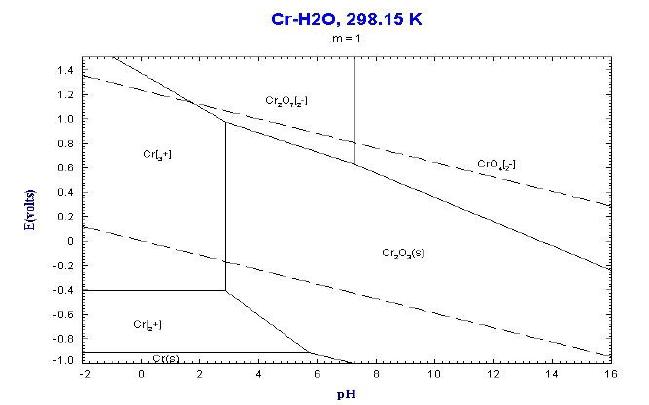
Wykres ten przedstawia obszary termodynamicznie trwałych form chromu przy założeniu, że stężenie formy jonowej wynosi 1 M. Na powietrzu pokrywa się warstwą pasywną Cr2O3. Traci odporność w środowisku kwaśnym oraz gdy ponieść temp. to także w zasadowym środowisku, podobnie jak Fe. Wysoka temperatura zwiększa także podatność na utlenienie do CrO42- w zasadowym środowisku.
Nikiel (symulacja):

Przy pH powyżej 4 nikiel nie koroduje w reakcji wypierania wodoru, ale pasywuje przy pH powyżej 6. Jest odporny na zasadowe środowisko.
Wiele związków organicznych jest inhibitorami korozji stali. Najwięcej to polarne substancje mające skłonność adsorpcji na wysokoenergetycznych powierzchniach. Szeroko stosowane są aminy. Czysta stal zawinięta w papier zaimpregnowany lotną aminą albo solą amoniową słabego kwasu jest chroniona przed korozją. Amin używa się w ogrzewaczach wody do zminimalizowania korozji. Powód skuteczności nie jest jasny. Mogą działać jako inhibitory bo są zasadami i mogą zobojętniać kwasy. Być może aminy silnie adsorbują się na powierzchni stali przez wiązania wodorowe lub tworzenie soli z kwasowymi miejscami na powierzchni stali. Ta zaadsorbowana warstwa może działać jako bariera chroniąca przed dostępem wody i tlenu do powierzchni stali. Jednakże ten mechanizm działa tylko w medium wodnym zawierającym inhibitor.
3) Ochrona przed korozją przez nieuszkodzone powłoki
Powłoki organiczne mogą być skutecznymi barierami chroniącymi stal gdy oczekuje się, że powłoka pokryje całą powierzchnię substratu i gdy pozostanie nienaruszona podczas użytkowania. Jednak kiedy oczekuje się, że powłoka nie pokryje całego substratu lub że zostanie naruszona podczas użytkowania, mogą być preferowane powłoki które mogą stłumić elektrochemiczne reakcje uwolnione podczas korozji.
Czynniki krytyczne
Przed rokiem 1950 n.e. sądzono, że powłoki chronią stal przed korozją działając jako bariera trzymająca wodę i tlen z dala od powierzchni stali. Wtedy zostało odkryte, że przepuszczalność powłok jest tak duża, że stężenie wody i tlenu przechodzących przez powłokę byłoby wyższe niż tempo zużywania w reakcji korozji niepokrytej stali. Zaproponowano zatem, że skuteczność powłoki związana jest z jej przewodnictwem. Rzeczywiście znaleziono korelację między skutecznością powłoki a jej przewodnictwem. Powłoki o dużej przewodności wykazują także dużą przepuszczalność wody.
Obecnie wiadomo, że skuteczność powłoki związana jest z jej adhezją w obecności wody. Funke zaproponował, że woda przenikająca przez nieuszkodzone powłoki może lokalnie powodować odczepienie powłoki od powierzchni stali. W takich przypadkach mówi się o słabej przyczepności na wilgotno. Woda i tlen rozpuszczony w wodzie mogą bezpośrednio kontaktować się z się z powierzchnią stali czego skutkiem jest zapoczątkowanie korozji. W trakcie korozji powstają jony Fe2+ i OH–, które prowadzą do utworzenia komórki osmotycznej. Ciśnienie osmotyczne generuje siły, które mogą odspajać więcej powłoki od substratu. Ciśnienie osmotyczne może osiągać wartości 2500 – 3000 kPa, podczas gdy odporność powłok organicznych na deformację jest niska, 6 – 40 kPa. Wtedy tworzy się pęcherz i rośnie, odsłaniając kolejne partie stali. Zaproponowano także nieosmotyczny mechanizm. Zakłada on, że woda zaabsorbowana przez powłokę powoduje występowanie napięć w płaszczyźnie powłoki i zwiększa wiązania pomiędzy powłoką a substratem stalowym elastycznie. W punkcie słabej adhezji powłoki do substratu wskutek naprężeń może dość do odspojenia. Udowodniono, że tempo wzrostu pęcherza jest zależne od modułu warstwy powłoki. Przy bardzo wysokich modułach wzrost pęcherza jest zminimalizowany.
Niezależnie od mechanizmu, kluczem do utrzymania ochrony przed korozją przez powłokę jest wystarczająca adhezja odporności na siły rozłączające. Każdy mechanizm mówi, że jeśli powłoka pokrywa całą powierzchnię stali w mikroskopowej skali tak samo dobrze jak w skali makroskopowej, jeśli adhezja na wilgotno może być osiągnięta na całej powierzchni, powłoka powinna definitywnie chronić przed korozją. Trudno jest spełnić oba te warunki nanosząc powłokę, wiec wysoki poziom adhezji na mokro jest ważny, ale nie jedynym czynnikiem wpływającym na ochronę przez powłokę. Na przykład wynikające z nano- i mikrochropowatości powierzchni metalu zagłębienia, które nie wszystkie mogą zostać wypełnione przez cząsteczki spoiwa z powodów sterycznych. Te niechronione jamki, chociaż małe, są wystarczająco duże by pozwolić na lokalne gromadzenie się wody i powstanie ogniwa korozyjnego. W dodatku do adhezji na mokro, odporność na korozję pozwala zwiększyć niska przenikalność dla wody i tlenu przez powłokę. W każdym razie jeśli adhezja na mokro jest kiepska, ochrona także jest słaba. Jeśli adhezja na mokro jest dobra to niskie tempo przenikania wody i tlenu może opóźniać straty adhezji wystarczająco długo by zapewnić odpowiednia ochronę przed korozją dla wielu praktycznych przypadków. Rysunek 16 przedstawia A – gładkie oddziaływania między powłoką a substratem, B – powierzchnia chropowata, C – chropowata powierzchnie z niecałkowitą penetracją jamek.

Adhezja – przygotowanie powierzchni
Pierwszym krokiem do osiągnięcia dobrej przyczepności jest oczyszczenie powierzchni przed malowaniem, szczególnie z soli i olejów. Naniesienie powłok fosforanowych przynosi dalsze korzyści. Gdy jest potrzebna dobra adhezja, ochrona przed korozją i względnie gładka powierzchnia trzeba obrobić metal chemicznie. Obróbka stali zwie się chemicznym przygotowaniem powierzchni lub powłokami konwersyjnymi. Stosowane są różne formy powłok konwersyjnych opartych na fosforanach. Takie powłoki są nanoszone poprzez spray lub zanurzenie w roztworze „fosforanu żelaza” w kwasie fosforowym. Ta metoda zapewnia łagodne trawienie powierzchni stali i powstawanie monowarstwy osadu fosforanu żelazawo-żelazowego. Adhezja powłoki znacząco wzrasta, ale ochrona przed korozją wzrasta tylko nieznacznie. Silniejszy wzrost ochrony przed korozją zapewnia zwykle użycie powłoki konwersyjnej fosforanu cynku. Stalowy obiekt jest zanurzony w r-rze fosforanu cynku w kwasie fosforowym i współstrącający się osad fosforanów żelaza i cynku formuje się na powierzchni stali. Adhezja i ochrona przed korozją rośnie wtedy znacznie. Koprecypitat formuje zazębiające się kryształy i przylegają szczelnie do powierzchni, zwiększając pole powierzchni stali w mikroskopowej skali. W zależności od stężenia cynku w kąpieli trawiącej różne kryształy mogą się odkładać. Przy względnie wysokich stężeniach cynku w kąpieli trawiącej tworzy się głównie hopeit Zn3(PO4)2*4H2O. W kąpielach o zubożonej zawartości cynku powstaje głównie fosfofilit Zn2Fe(PO4)2*4H2O. Proces wytrawiania w kąpieli cynkowej przedstawia rysunek 17.

Rys. 15
Skuteczność powłoki konwersyjnej jest określona przez jednolitość i stopień wytrawienia powierzchni. Powłoki fosforanu cynku są generalnie nanoszone w granicach 1,5 – 4,5 g/m2. Są stosowane także inne powłoki fosforanowe od wspomnianych. Reakcje na rysunku 15 są proste, ale osiągnięcie wymaganego szybkiego tempa reakcji pozwalającego na minimalne czasy trawienia jest trudniejsze. Własne receptury skracają czas wytrawiania do rzędu minut lub sekund. Jakość trawienia zależy od czasu, temperatury i pH. Te i inne zmienne muszą być ściśle kontrolowane dla pewności, że pożądany typ i rozmiary kryształów są formowane. Trawiona powierzchnia musi być dokładnie wypłukana celem usunięcia wszelakich soli mogących prowadzić do powstania pęcherzy gdy para wodna przenika przez powłokę, płukanie usuwa także luźno związane kryształy. Do ostatniego płukania stosowano kąpiel zawierającą małe stężenie kwasu chromowego dla ochrony przed korozją. Jednak z powodów zagrożenia toksycznym chromem na szóstym stopniu utlenienia kąpiele te zastępuje się innymi. Nad fosforanem cynku, kąpiel 0,5% metylotrimetoksysilanu z odpowiednim stężeniem H2ZrF6 daje pH ok. 4 i wyniki wskazują, że daje lepsze rezultaty niż kąpiel chromianowa. Jednakże nad fosforanem żelaza lepsze wyniki daje kąpiel chromianowa.
Na podstawie:
Organic Coatings. Science and Technology; Z. W. Wicks, F. N. Jones, S. P. Papas, D. A. Wicks; Wiley 2007
Rysunki zaczerpnięto także z czasopisma PPCJ