
Wśród efektownych reakcji chemicznych niewątpliwie silną pozycję zajmują reakcje luminescencyjne. Na ogół doświadczenia z chemiluminescencją wymagają użycia drogich albo żmudnych w przygotowaniu substancji. Przykładem jest luminol, opisany chyba w każdym zbiorze z doświadczeniami. W domowych warunkach otrzymywanie luminolu jest czasochłonne i dość problematyczne, z powodu uciążliwej reakcji nitrowania kwasu lub bezwodnika ftalowego. Najprościej taki luminol po prostu kupić, co dzisiaj nie jest dużym problemem. Jednak poza luminolem potrzebne są jeszcze inne substancje do wykonania doświadczeń. Innymi przykładami substancji używanych do doświadczeń chemiluminescencyjnych są szczawiany podstawionych fenyli, chlorek oksalilu czy lucygenina. Otrzymywanie ich w domowych warunkach także nastręcza trudności. Jednakże jak każdy chemik-praktyk, zdaję sobie sprawę z tego, że odczynnik kupiony to nie to samo, co odczynnik otrzymany samemu. Z tego powodu postanowiłem się skupić na lofinie, której metody otrzymywania są względnie proste w domowych warunkach. To samo dotyczy demonstracji jej właściwości luminescencyjnych. W tym artykule opiszę metodę prostszą oraz bardziej zaawansowaną.
Nasza główna bohaterka została odkryta w roku 1877 przez jednego z najwybitniejszych polskich chemików organików XIX stulecia, Bronisława Radziszewskiego (1838 – 1914), którego możemy zobaczyć poniżej.
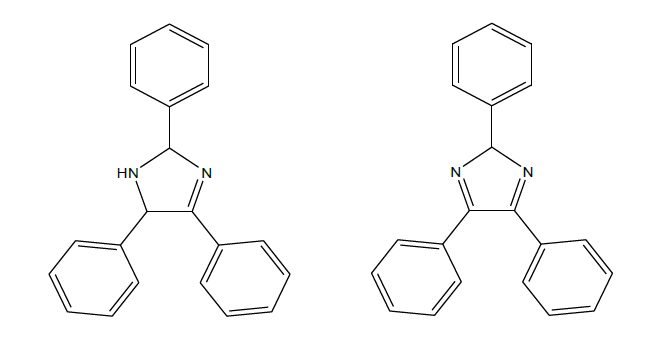
Z chemicznego punktu widzenia lofina jest 2,4,5-trifenyloimidazolem. Jej strukturę widzimy na rysunku 1.

Już sama nazwa wskazuje, że jest to związek heterocykliczny, szkielet imidazolowy zawiera bowiem 2 atomy azotu. To właśnie dzięki nim zawdzięcza ona swoje luminescencyjne właściwości, jeden atom azotu posiada względnie luźno związany wodoru, czyli proton o kwasowych właściwościach, ale dzięki temu, że łatwo może oddysocjować, ten atom azotu najchętniej ulega reakcjom. Drugi atom azotu wykazuje za to właściwości zasadowe. Można sobie tak rozrysować struktury rezonansowe imidazolu, by samemu wskazać taką sytuację. Z powyższego wynika, że lofina, jako imidazol, jest amfoteryczna. Jedną z pierwszych metod otrzymywania wielu związków było ogrzewanie substratów w obecności tlenu atmosferycznego, tak samo postąpił Radziszewski. Użył on znanego już wcześniej hydrobenzamidu, który ogrzewał do wysokich temperatur w parownicy w obecności powietrza. Rys. 2 pokazuje cykl przemian od hydrobenzamidu do lofiny.

Warto wspomnieć, że sam Radziszewski początkowo zaproponował inne wzory strukturalne dla otrzymanej przez siebie substancji, które są pokazane na rys. 3. Widać różnicę pomiędzy rzeczywistą strukturą lofiny a hipotetycznymi Radziszewskiego? Dodam także, że Radziszewski kilka lat po otrzymaniu lofiny tą drogą, zbadał także inną metodę otrzymywania imidazoli, znaną jako reakcja Radziszewskiego. Ta druga metoda jest opisana także w tym artykule, jako metoda bardziej zaawansowana. Pierścień imidazolowy został syntetycznie otrzymany już w roku 1840, ale dopiero Radziszewski w 40 lat później zbadał tę reakcję, która została nazwana jego nazwiskiem.
Hydrobenzamid, mimo tajemniczej nazwy, jest związkiem o dość prostej strukturze i bardzo łatwy w otrzymaniu. Równanie jego otrzymywania przedstawia rysunek 4.

Struktura tego związku była znana już swemu odkrywcy, Aleksandrowi Borodinowi. Jego nazwisko zapisało się w historii głównie jako kompozytor, a nie jako chemik, mimo, że także był zdolnym organikiem. Osobiście Borodin ubolewał z tego powodu, że nie jest znany jako chemik, tylko jako kompozytor, poza muzyką był także doktorem nauk chemicznych. Znany go przede wszystkim jako twórcę opery Książę Igor. Spora część z nas zapewne kojarzy ze słyszenia arię tej opery:
[youtube http://www.youtube.com/watch?v=WahcTznCRm4&version=3&feature=player_detailpage]
Fotografia poniżej pokazuje Tego kompozytora.

Tymczasem wróćmy do chemii. Hydrobenzamid to nieco pechowa nazwa. Przede wszystkim jak wynika ze wzoru strukturalnego, nie jest to amid, tylko imina. Dawniej chemicy dodawali przedrostek hydro, uwzględniając w ten sposób, że daną grupę funkcyjną można otrzymac przez redukcję wodorem innej grupy, tak wyłoniono grupę hydramidów, produktów częściowej redukcji amidów. Dzisiaj wiemy, że takie związki są po prostu iminami. Hydrobenzamid, bo związek taki możnaby otrzymać przez redukcję benzamidu, czyli amidu kwasu benzoesowego. Jednakże w literaturze można spotkać także nazwę tribenzylidenodiamina. Ta nazwa jestnak jest nieścisła, gdyż w cząsteczce tej mamy typowe grupy iminowe, stąd osobiście używam skorygowanej nazwy: tribenzylidenodiimina. Systematyczna nazwa tego związku to 1,3,5-trifenylo-2,4-diaza-1,4-pentadien. W opracowaniu tym podam także 2 przepisy na hydrobenzamid, w wersji nieco prostszej i nieco bardziej zaawansowanej, ale obie metody mogą być z powodzeniem zastosowane w domu, tak samo jak i przetworzenie go na lofinę.
Hydrobenzamid. Metoda z użyciem wody amoniakalnej
Odczynniki:
aldehyd benzoesowy
woda amoniakalna (najlepiej 25%)
woda destylowana lub demineralizowana
izopropanol lub etanol (do krystalizacji)
Sprzęt:
butla szklana lub kolba stożkowa ze szlifem lub dopasowanym korkiem
lejek Buechnera
pompka próżniowa
szalka Petriego
chłodnica powietrzna, kulkowa lub rurka szklana
bibuła filtracyjna jakościowa
Aldehyd benzoesowy najlepiej, jeśli jest świeżo przedestylowany, że względu na jego łatwość utleniania na powietrzu. Wystarczy go poddać prostej destylacji, zbierając frakcję wrzącą pomiędzy 178-180 oC. Czystość aldehydu na znaczący wpływ na ilość i czystość otrzymanego produktu. Aldehyd benzoesowy, jeśli jest kupiony u solidnych dostawców, jest bezbarwny i nie wymaga wtedy destylowania. Jeśli jest już żółty to znacznie odbije się na jakości produktu, który koniecznie trzeba będzie oczyszczać. Aldehyd, który przybrał już barwę brązową zawiera już duże ilości produktów utleniania i innych reakcji i nie nadaje się do użycia bez oczyszczania. Jeśli nie mamy możliwości destylacji, usuńmy z niego chociaż kwas benzoesowy przez wytrząsanie zimnego aldehydu z 5% r-rem NaOH lub KOH a następnie co najmniej 3 razy wodą. Pozostałości wody w aldehydzie nie będą nam przeszkadzać. Ważne, aby nie narażać aldehydu na dłuższy kontakt z powietrzem. Aldehyd, w ilości ok. 10 g zamykamy w butli lub kolbie z 20-30 ml wody amoniakalnej. Całość odstawiamy na 2 doby. W tym czasie aldehyd powinien całkowicie przereagować a na dnie powinna się zebrać spora ilość białego osadu. Jest to hydrobenzamid. Zbrylony osad rozbijamy bagietką lub szpatułką. Fazę wodną usuwamy z naczynia reakcyjnego i zalewamy produkt wodą i wytrząsamy, celem wypłukania resztek amoniaku. Czynność kilka razy można powtórzyć. Przemywanie wodą można wykonać na lejku Buechnera, o ile mamy warunki do sączenia próżniowego. Na sam koniec można przemyć 10-20% r-rem etanolu lub izopropanolu. Osad przenosimy na krążek bibuły umieszczony na szalce Petriego i suszymy w temperaturze pokojowej lub nieco wyższej, ale nie ponad 60 oC. Nie należy za bardzo ogrzewać wilgotnego hydrobenzamidu, gdyż spowoduje to jego hydrolizę do aldehydu benzoesowego. Jeśli aldehyd był czysty, to osad ten jest też na tyle czysty, że nie powinien wymagać dalszego oczyszczania przez krystalizację. Czysty topi się w 100-101 oC. Jesli jego kolor jak i kolor wody amoniakalnej po reakcji świadczą o zanieczyszczeniach produktu, pochodzących z aldehydu benzoesowego, to konieczna będzie krystalizacja. W tym celu suchy hydrobenzamid ogrzewamy w kolbce kulistej z izopropanolem lub etanolem (może być spirytus). Bardzo wydajna jest krystalizacja z izopropanolu. Początkowo należy na jeden gram osadu brać 1 ml alkoholu i w razie potrzeby dodawać tyle tyle, aby całkowicie rozpuścić produkt w temperaturze wrzenia. Za chłodnicę wystarczy kawałek pionowej rurki (chłodnica powietrzna), gdyż i tak ogrzewamy tylko do domentu rozpuszczenia substancji. Po ochłodzeniu i wykrystalizowaniu produktu należy go odsączyć i przepłukać niewielką ilością zimnego alkoholu. Po wysuszeniu produkt jest gotowy do dalszego użycia. Wydajność do ok. 90%
Hydrobenzamid. Metoda z użyciem chlorku amonu
Odczynniki:
benzaldehyd
chlorek amonu
wodorotlenek sodu (lub potasu)
woda amoniakalna
izopropanol
woda destylowana lub demineralizowana
Sprzęt
kolba stożkowa lub butla szklana
lejek Buechnera lub ze spiekiem
cylinder miarowy
pompka próżniowa
Tak jak poprzednio najlepiej użyć aldehydu czystego. Hydrobenzamid otrzymany tą metodą z czystego aldehydu gwarantuje wydajność niemal 100% i powstały produkt jest czysty (tt 100-101 oC). Najpierw przygotowujemy odczynnik amoniakalny: do naczynia reakcyjnego wlewamy 18 ml wody destylowanej i rozpuszczamy w niej chlorek amonu (3,8 g). Następnie dodajemy wodorotlenku sodu (0,8 g) i także rozpuszczamy, a na koniec dodajemy 6 ml wody amoniakalnej. Aldehyd (10,6 g)rozpuszczamy w izopropanolu (11 ml) i wlewamy całość do gotowego odczynnika amoniakalnego. Naczynie zamykamy i odstawiamy na 24 godziny. Jeśli aldehyd nie był czysty, to początkowo wypada żółty olej zamiast osadu, który następnie ulega zestaleniu. W przypadku, gdyby powstajacy osad utworzył skorupę oddzielającą fazę wodną od aldehydu, należy go rozbić. Po co najmniej 12 h w temperaturze pokojowej wytrącony osad rozbijamy bagietką lub łyżeczką i przenosimy na lejek Buechnera. W przypadku braku lejka osad delikatnie dekantujemy znad fazy wodnej. Osad na lejku lub w kolbie płuczemy kilka razy wodą destylowaną. Dobre odmycie osadu można rozpoznać po braku obecności chlorków w zakwaszonym przesączu w próbie na chlorki z r-rem azotanu srebra, ale nie jest to potrzebne. Papierkiem można też sprawdzić odczyn przesączu, jeśli jest obojętny, to osad też można uznać za dobrze odmyty od resztek soli. Niewielki dodatek izopropanolu do wody poprawia zwilżalność i skuteczność przemywania osadu. Wystarczy ok. 5% alkoholu, zbyt duże ilości mogą zacząć rozpuszczać osad. Jeśli nie mamy lejka, to wszystkie te operacje można zastąpić wytrząsaniem osadu w kolbie, ale jest to żmudniejsze i wymaga delikatnego zlewania wody znad osadu. wypłukany osad suszymy tak jak poprzednio. Jeśli użyty aldehyd nie był czysty, produkt można krystalizować tak jak poprzednio. Przed dalszym użyciem produkt dokładnie wysuszyć. Przy przestrzeganiu prawidłowych operacji wydajność przekracza 99% (8,9 g). Wydajność krystalizacji czystego związku z alkoholu izopropylowego wynosi 94%.
Lofina. Metoda historyczna (Radziszewskiego)
Odczynniki:
Hydrobenzamid
Sprzęt:
parownica
bagietka szklana
czasza grzejna lub palnik
trójnóg lub statyw z kółkiem do tygli
Uwaga, w reakcji tej powstaje nieco drażniących par. Najlepiej wykonywać ją pod wyciągiem lub w dobrze wentylowanym pomieszczeniu. Operację najlepiej wykonać na czaszy grzejnej. Wtedy nie potrzebujemy nic poza parownicą i bagietką. Hydrobenzamid umieszczamy w parownicy, którą silnie ogrzewamy. Po stopieniu się substrat zaczyna ciemnieć a następnie dymić. W tym czasie musimy intensywnie mieszać bagietką stop. Po kilku minutach przerwać ogrzewanie i zostawić parownicę do ostygnięcia. Szklistą masę wyjąć z parownicy, pokruszyć np. w moździerzu i przenieść do kolbki kulistej, zalać metanolem lub innym alkoholem (izopropylowym, etanolem). Ilość alkoholu potrzebną do całkowitego rozpuszczenia stopu na gorąco trzeba ustalić doświadczalnie. Po całkowitym rozpuszczeniu należy odstawić kolbę do ostygnięcia i wykrystalizowania lofiny, która jest drobnym osadem. Odsączamy ją pod próżnią i przemywamy zimnym alkoholem. Wydajność 20-30%. Tak otrzymana lofina jest wystarczającej jakości do prób chemiluminescencji.
Lofina. Metoda preparatywna i wielkolaboratoryjna wg reakcji Radziszewskiego

Odczynniki:
aldehyd benzoesowy
benzil (dibenzoil)
octan amonu
kwas octowy lodowaty
woda destylowana
metanol lub inny alkohol
Sprzęt:
kolba kulista 500 ml
chłodnica zwrotna
czasza grzejna
statyw
małe wiaderko plastikowe
mieszadło laboratoryjne lub mikser
Do kolby kulistej wprowadzamy ok. 250 ml lodowatego kwasu octowego, 80 g octanu amonu, 9 g aldehydu benzoesowego, 18 g benzilu oraz kilka zarodników wrzenia. Ogrzewamy do wrzenia przez 3 godziny pod chłodnicą zwrotną. Po ostygnięciu mieszaninę poreakcyjną wlewamy cienkim strumieniem do 2 L intensywnie mieszanej wody destylowanej, wprost na łopatki mieszadła lub na widełki miksera. Zbyt słabo mieszana woda powoduje postanie brył, które trudno potem wypłukać z resztek kwasu i soli. Wytrącony osad przenieść na lejek Buechnera i po odsączeniu osad wypłukać wodą destylowaną. Odmyty od kwasu osad wysuszyć. Przekrystalizować z metanolu lub innego alkoholu. Na 1 g osadu brać ok. 40 ml metanolu i w razie potrzeby dodać więcej. W przypadku innego alkoholu ustalić niezbędną ilość doświadczalnie. Po ostygnięciu pozostawić do krystalizacji na 24 godziny. Wydajność krystalizacji ok. 80% z metanolu, ale można ją nieco zwiększyć umieszczając r-r w zamrażarce i dopiero po pełnej krystalizacji przesączyć. Otrzymuje się ok. 20 g lofiny o tt. 276-278 oC.
Demonstracja reakcji luminescencyjnej lofiny
Odczynniki:
lofina
woda utleniona lub perhydrol (może być nawet wybielacz na bazie nadtlenku wodoru)
r-r podchlorynu sodu o zawartości co najmniej 4% chloru (najlepszy jest tani wybielacz Bielinek)
alkohol etylowy lub metylowy (ew. denaturat)
wodorotlenek potasu
Przygotowujemy sobie 4 roztwory:
A: 0,2 g lofiny w 10 ml alkoholu
B: ok. 0,5 ml perhydrolu lub 3 ml wody utlenionej uzupełnić alkoholem do objętości 25 ml
C: 1 g wodorotlenku potasu rozpuszczamy w 15-20 ml wody i uzupełniamy alkoholem do 25 ml
D: 2,5 ml r-ru podchlorynu uzupełnić wodą do 25 ml.
W wysokiej zlewce lub cylindrze mieszamy ze sobą r-ry A, B, i C. Następnie, w ciemny pomieszczeniu, wlewamy r-r D. Pojawia się żółta luminescencja, trwająca kilkanaście sekund oraz powstają pęcherzyki gazu (tlen). Dodatek kolejnych porcji wybielacza wznawia luminescencję kilka razy.
Z moich własnych obserwacji wynika, że intensywniej świeci p,p’,p”-trimetylolofina. Otrzymać ją można stosując zamiast aldehydu benzoesowego aldehyd p-tolilowy (p-metylobenzoesowy). Aldehyd ten reaguje jednak znacznie wolniej z amoniakiem i trzeba poczekać około tygodnia na powstanie odpowiedniej ilości hydrobenzamidu.
Wydajność jego jest jednak niższa, rzędu 90%, a temperatura topnienia wynosi 95-96 oC. Isnieją też metody otrzymywania lofiny innymi metodami, ale o nich napiszę, gdy zostaną przez mnie zweryfikowane.
Witam
Czy sączenie próżniowe jest niezbędne do otrzymania substancji pierwszym sposobem?
Witam, sączenie próżniowe zawsze bardzo ułatwia pracę. Nie jest niezbędne, jednakże produkt będzie wymagał dłuższego czasu suszenia i będzie gorszej czystości.
Miałbym jeszcze jedno pytanie (przy okazji dziękuję za odpowiedź). Gdzie można poczytać o sączeniu próżniowym?
Polecam preparatykę organiczną lub nieorganiczną, każda zawiera opisy technik laboratoryjnych.