Wiązanie koordynacyjne – fakt czy mit?
UWAGA! Czytelników zachęcam do własnoręcznego rozrysowania poruszonych przypadków elektronowo.
Dziś poruszymy problem często spotykany: wiązanie koordynacyjne. Czym ono właściwie jest? Czy ono istnieje? Tak, celowo zadaję pytanie o jego istnienie, bo jest jedno z problematycznych zagadnień edukacji chemicznej i często spotyka się niewłaściwie stwierdzenia na jego temat nie tylko wśród osób uczących się chemii, co i w szkolnych materiałach edukacyjnych albo i wśród osób odpowiedzialnych za edukację. Zacznijmy jednak od definicji wiązania koordynacyjnego:
Takie wiązanie, w którym wiążąca para elektronowa pochodzi od jednego atomu.
W bardziej „typowym” przypadku wiązanie powstaje przez uwspólnienie elektronów pochodzących od obu łączących się atomów: każdy daje po jednym i jest po równo – powstaje wiążąca para, czyli wiązanie chemiczne. Z definicji wiązania koordynacyjnego wynika, że jest to wiązanie kowalencyjne – nie może być inaczej, gdyż w przypadku wiązania jonowego nie ma mowy o parach wiążących – w związkach jonowych rozpatrujemy tylko jony – jeden jon ma za dużo elektronów, a drugi za mało. Jeden jest anionem, a drugi kationem. Jeden zyskał, drugi stracił, a sumaryczny ładunek jest równy zero. Jest to przy okazji odpowiedź na jedno z częściej spotykanych pytań o charakter wiązania koordynacyjnego: jest to wiązanie kowalencyjne z definicji. Wiązania kowalencyjne możemy rozpatrywać ze względu na trzy kryteria:
– sposobu nakładania się sfer walencyjnych
– polarności wiązania (różnicę elektroujemności wiążących się atomów)
– pochodzenia wiążących elektronów
Jeśli rozpatrujemy wiązania ze względu na pierwsze kryterium, to dzielimy je na sigma (σ) i pi (π), jeśli ze względu na drugie, to na spolaryzowane i niespolaryzowane. Jeśli z kolei na pochodzenie elektronów – to na koordynacyjne i niekoordynacyjne, czyli „zwykłe”. Często popełnianym przez niektórych błędem jest podział wiązań na kowalencyjne i koordynacyjne – podczas gdy to samo wiązanie może być zarówno koordynacyjne jak i kowalencyjne spolaryzowane równocześnie – bo za pierwszym razem rozpatrujemy je z perspektywy pochodzenia elektronów, a za drugim to ze względu na różnicę elektroujemności.
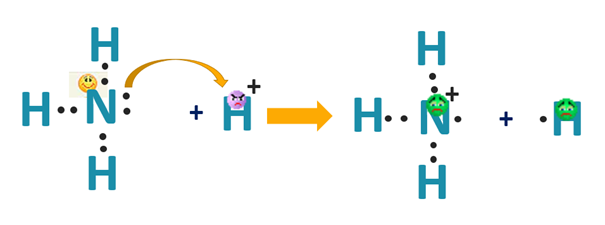
Nazwałem wiązanie powstające przez uwspólnienie niesparowanych elektronów zwykłym, bo wręcz odruchowo zakładamy, że jeśli coś się ze sobą wiąże, to uwspólnia niesparowane elektrony i atomy dają taką samą liczbę elektronów do utworzenia wiązania lub wiązań. Tak nas przyzwyczaja po prostu edukacja. Ale czy zawsze tak jest? Za moment się przekonamy. W szkole o wiązaniu koordynacyjnym pierwszy raz się dowiadujemy często przy okazji omawiania jonu amonowego. Wolny atom azotu ma 3 niesparowane elektrony walencyjne, oraz tzw. wolną parę elektronową. Z kolei wolny atom wodoru ma jeden niesparowany elektron. Wynika z tego, że atomy tych pierwiastków przereagują ze sobą w stosunku 1:3 i dadzą cząsteczkę amoniaku:

Teraz pary elektronowe zastępujemy kreską i otrzymujemy:

Atom azotu ma teraz 8 elektronów na powłoce walencyjnej, czyli osiągnął konfigurację neonu, a atom wodoru ma dwa, czyli osiągnął konfigurację helu. Nie ma miejsca na czwarty atom wodoru ze swoim kolejnym niesparowanym elektronem – bo azot ma już zapełnioną całą powłokę walencyjną. Ale co by się stało, gdyby do cząsteczki amoniaku zbliżył się kation wodorowy, czyli proton H+? Proton nie ma elektronu, więc nie spowoduje, że atom azotu będzie miał 9 elektronów w sferze walencyjnej, a sam proton dąży do konfiguracji helu. Szuka więc pary elektronowej. Tymczasem azot, mimo związania się z trzema atomami wodoru, nadal ma wolną parę. Gdyby więc wykorzystać tę parę do utworzenia wiązania? Nie ma miejsca na kolejny obojętny atom wodoru, ale jest miejsce na kation wodoru. Atom azotu staje się więc dostawcą elektronów w tworzącym się nowym wiązaniu kowalencyjnym.

To czwarte wiązanie zwiemy koordynacyjnym. We wzorach strukturalnych możemy uwzględnić wiązanie koordynacyjne na dwa sposoby: przez uwzględnienie ładunków formalnych, albo strzałką:

Ładunek dodatni na atomie azotu bierze się stąd, że donor wiążącej pary formalnie traci elektron. Pamiętajmy, że jon ten powstał w reakcji amoniaku z H+, a ładunek nie znika, jest taki sam po stronie substratów, jak i produktów. Tyle że przed reakcją wodór w ogóle nie posiadał elektronu, podczas gdy teraz ma do dyspozycji wspólną parę – wzbogacił się o elektrony, a atom azotu z kolei stracił. Jeśli do kogoś to nie przemawia, to tworzenie wiązania koordynacyjnego można sobie wyobrazić w ten sposób: najpierw atom azotu oddaje protonowi jeden z dostępnych elektronów. Sam wtedy staje się kationem z niesparowanym elektronem (rodnikokationem):
Ale obydwa atomy nie spełniają teraz reguły helowca. Następnie powstałe rodniki tworzą wiązanie pomiędzy sobą uwspólniając swoje niesparowane elektrony:

Oba atomy mają teraz konfigurację helowca, atom wodoru zyskał elektron (wcześnie w ogóle nie miał elektronów), a atom azotu formalnie stracić elektron – dlatego ładunek dodatni w jonie amonowym jest właśnie zgromadzony na atomie azotu, a nie na protonie, który przecież przestał być protonem (H+) w wyniku tej reakcji! Pozostaje odpowiedzieć na kolejne pytanie: czy to wiązanie różni się od trzech pozostałych? Na rysunku, na którym zaznaczyliśmy kolorowymi kropkami elektrony walencyjne możemy wskazać, które wiązanie jest koordynacyjne. Lecz czy w rzeczywistym jonie amonowym możemy wskazać, które z wiązań N-H jest koordynacyjnym? Niestety nie – nie jesteśmy w stanie tego wskazać, bo wszystkie wiązania są identyczne. Identyczne, bo zbudowane z takich samych elektronów i takich samych atomów, jak pozostałe. Wynika to stąd, że atomy wszystkich pierwiastków chemicznych zbudowane są z identycznych cząstek elementarnych, a różnią się jedynie ich liczbą. Nie ma znaczenia, czy to atom azotu, tlenu, czy wodoru – każdy zawiera identyczne elektrony, którymi zresztą mogą się atomy wymieniać. Co innego, gdyby atomy były zbudowane z różnych cząstek, np. elektrony azotu byłyby większe i cięższe inne niż elektrony wodoru. Wtedy wiązanie powstałe przez uwspólnienie pojedynczych elektronów byłoby inne, niż to powstałe przez dostarczenie pary przez jeden z atomów. Załóżmy, że elektron wodoru waży 1, a elektron azotu 4 jednostki. Wtedy każde wiązanie N-H w amoniaku ważyłoby 5 jednostek masy, a wolna para elektronowa ważyłaby 8 jednostek. Gdyby ta para utworzyła wiązanie ze zbliżającym się protonem, to powstałe wiązanie różniło by się od reszty, bo byłoby zbudowane z innych elektronów i moglibyśmy wskazać, że różni się od reszty masą, długością i energią. Lecz niestety cząstki elementarne budujące wszystkie atomy są identyczne, a atomy poszczególnych pierwiastków różnią się tylko ich liczbą – nie jesteśmy w stanie stwierdzić, które konkretnie wiązanie w jonie amonowym jest koordynacyjne i stwierdzamy co najwyżej, że ono jest. Co więcej, to prawdopodobieństwo, że któryś z protonów oddysocjuje także jest identyczne i wynosi 25%, czyli jedną czwartą. Oznacza to, że gdybyśmy potrafili na atomach wodoru narysować numerki i oznaczyli atomy wodoru w cząsteczce NH3 cyframi 1, 2, 3, a przyłączony proton otrzymałby numer 4, to szansa, że w wyniku dysocjacji odejdzie proton o numerze 4 wynosi 25%, a pozostałe (1, 2, 3) mają identyczną szansę, bo przecież są z nim identyczne. Takie atomy nazywamy równocennymi. Amoniak, zarówno w postaci gazowej jak i wodnego roztworu (wody amoniakalnej), reaguje z kwasami protonowymi dając sole amonowe, które łatwo wydzielić przez odparowanie wody z mieszaniny poreakcyjnej. Reaguje także z gazowymi halogenowodorami, np. z gazowym chlorowodorem, tworząc charakterystyczną mgłę chlorku amonu:

Chlorek amonu jest związkiem jonowym, zbudowanym z kompleksowego kationu amonowego NH4+ i przeciwjonu Cl–, który równoważy jego ładunek dodatni. Elektrostatyczne przyciąganie się tych jonów zwiemy wiązaniem jonowym. Analogiczna sytuacja jak w jonie amoniowym występuje w jonie hydroniowym H3O+.

W klasycznych reakcjach chemicznych powstaje on przez utworzenie wiązania koordynacyjnego pomiędzy atomem tlenu z cząsteczki wody, a kationem wodorowym, czyli protonem – analogicznie jak można otrzymać jon amonowy:

I podobnie jak w poprzednim przypadku – wiązanie to jest nierozróżnialne od dwóch pozostałych i ma takie same właściwości. Powstało tylko w innym sposób, niż można było utworzyć dwa pozostałe w cząsteczce wody. Lecz czy na pewno? Wrócimy jeszcze do tego przypadku.
Kolejny raz z wiązaniem koordynacyjnym spotykamy się chociażby w przypadku tlenku siarki(IV), gdy znamy już regułę oktetu i okazuje się, że utworzenie dwóch wiązań podwójnych S=O jest niezgodne z tą regułą, gdyż wtedy siarka miałaby o dwa elektrony za dużo i nie osiągnęłaby konfiguracji helowca. Dwutlenek siarki jest związkiem polarnym i ma budowę kątową.

Zadamy sobie zatem pytanie, czy w dwutlenku siarki wiązanie podwójne różni się od koordynacyjnego? Czy jesteśmy w stanie odróżnić od siebie te dwa wiązania? Tutaj już by się wydawało, że tak, bo przecież jedno jest podwójne, a drugie pojedyncze, więc powinny się różnić długością i energią. Lecz niestety jesteśmy w błędzie – i tutaj wiązania są równocenne. Wynika to z faktu, że w takich układach, gdzie wiązanie podwójne sąsiaduje z atomem z wolną parą elektronową następuje sprzężenie i zjawisko rezonansu elektronów:

Elektrony są w ciągłym ruchu i wiązanie podwójne przechodzi w pojedyncze i na odwrót. Powoduje to, że w rzeczywistości wiązania w tlenku siarki mają charakter pośredni pomiędzy pojedynczym, a podwójnym i są tej samej długości: 143 pm. Zauważmy jeszcze coś: jeśli podwójne powstało przez uwspólnienie niesparowanych elektronów atomów siarki i tlenu, to czy w chwili, w której jedna z wiążących par przeskoczyła na tlen – powstałe wiązanie pojedyncze stało się nagle koordynacyjnym? Logika wskazuje, że nie, ale formalnie zakładamy, że to wiązanie jest teraz koordynacyjne. A z drugiej strony mamy odwrotną sytuację – formalnie wiązanie koordynacyjne przestało być koordynacyjnym, mimo, że wiążącym elektronom nadal można przypisać pochodzenie od atomu siarki!
Jeszcze ciekawiej sytuacja wygląda w takich związkach jak SO3 czy H2SO4, gdzie trzymając się reguły oktetu, możemy utworzyć aż dwa wiązania koordynacyjne. Wiązanie koordynacyjne i rezonans możemy też rozpatrywać w przypadku kwasu azotowego(V) i jego soli. O ile w przypadku pierwiastków drugiego okresu, gdzie należy azot, wiązanie koordynacyjne w cząsteczce HNO3 jest wymuszone ograniczoną pojemnością powłoki walencyjnej, o tyle w okresie następnym, gdzie należą fosfor i siarka, jest to już założenie związane ze stosowaniem reguły oktetu, gdyż ze względu na większą pojemność powłoki walencyjnej, od okresu trzeciego począwszy obserwuje się liczne odstępstwa od tej reguły. I tak w kwasie siarkowym(VI) czy w jego bezwodniku (SO3) istnienie dwóch wiązań podwójnych jest bardziej korzystne energetycznie, niż dwóch wiązań koordynacyjnych. Trzymając się reguły oktetu, to nie mogą istnieć takie związki jak NCl5, PCl5, SF6, H5IO6 – i rzeczywiście: nie istnieje NCl5, ale ISTNIEJĄ pozostałe związki. Trzymając się reguły oktetu nie można rozrysować ich wzorów elektronowych, ale biorąc pod uwagę większą pojemność powłoki walencyjnej siarki, fosforu czy jodu – jak najbardziej można uzasadnić ich istnienie. Jedynie azot jest całkowicie ograniczony regułą oktetu, bo jego sfera walencyjna mieści maksymalnie 8 elektronów.
Ze względu na sposób nakładania się orbitali wiązania dzielimy na typu σ i typu π – wiązania koordynacyjne też mogą powstawać zarówno przez czołowe, jak i boczne nakładanie się orbitali, czyli być typu s i p, co jak już mówiłem wynika z tego, że są to różne kryteria rozpatrywania wiązań. Przykładem związku, gdzie wiązanie koordynacyjne należy do typu p jest tlenek węgla(II) CO.

Choć oczywiście możemy dyskutować o tym, które z wiązań jest koordynacyjne: czy to jedno z wiązań typu π, czy też to typu σ? Jeśli dociekliwi czytelnicy spróbują rozrysować elektronowo tworzenie wiązania pomiędzy atomem tlenu i atomem węgla, to można zacząć od utworzenie wiązania koordynacyjnego, a dwa następne przez uwspólnienie niesparowanych elektronów, które będą wiązaniami π. Powszechnie jednak przyjmuje się, że wiązanie koordynacyjne w cząsteczce CO jest typu π, zaiste ze względu na rezonans w cząsteczce CO – teoria zakłada, że za rezonans odpowiadają nie uczestniczące w hybrydyzacji orbitale typu p. Ze względu na rezonans dominującą formą w tlenku węgla(II) jest ta widoczna na rysunku – forma z wiązaniem podwójnym stanowi ok. 20%.
Innym przykładem jest reakcja tworzenia ditiortęcianów(II):

Gdzie anion siarczkowy tworzy aż dwa wiązania koordynacyjne z atomem rtęci. Jedno z nich jest typu σ, a drugie typu π. W tym przypadku już nie takich wątpliwości, jak w przypadku CO.
Teraz poruszymy następną kwestię. Wiemy już, że wiązanie koordynacyjne w takich jonach jak amonowy czy hydroniowy jest nierozróżnialne od reszty i dowolne wiązanie możemy sobie wybrać i określić, że akurat to wiązanie jest koordynacyjnym – i nikt nie ma prawa zakwestionować naszego wyboru. Co więcej, to to samo wiązanie kowalencyjne możemy rozpatrywać zarówno jako koordynacyjne jak i jako „zwykłe”. Przykład? Proszę bardzo. Cząsteczka wody zawiera 2 wiązania H-O. Wiązanie to może zostać rozerwane (zdysocjować) na dwa sposoby:
– symetrycznie, powstają obojętne atomy i cząstki, a wiążące elektrony są dzielone symetrycznie: po jednym na atom. W wyniku rozpadu wiązania H-O w cząsteczce wody powstają dwa rodniki:

Taki rozpad nazywany homolitycznym, a często także dysocjacją termiczną.
– niesymetrycznie, jeden ze związanych atomów zabiera całą parę wiążącą i powstają cząstki obdarowane ładunkiem elektrycznym, czyli jony H+ i OH–:

Taki rozpad zwiemy heterolitycznym lub dysocjacją elektrolityczną.
Wodę można otrzymać w reakcji pierwiastków chemicznych ze sobą, przez spalanie wodoru w tlenie, wtedy elektrony pochodzą od obu atomów. Można rozpatrywać tworzenie wiązań następująco:

Ale woda może powstać też w reakcji rodników ze sobą – w procesie odwrotnym do zapisanego poprzednio:

Tutaj także oba atomy dostarczają równą ilość elektronów do powstającego wiązania. Jednakże wodę możemy także otrzymać w reakcji wodorotlenku z kwasem, czyli w reakcji zobojętniania:

Co elektronowo możemy zapisać jako (kolor czerwony oznacza ładunek jonu):

I tutaj niespodzianka, bo tlen jest donorem pary wiążącej, czyli utworzone wiązanie H-O jest wiązaniem koordynacyjnym! Jeśli pójdziemy o poziom wyżej, to możemy rozpatrzeć reakcję tlenku zasadowego, czyli zbudowanego z jonów O2- z kwasem (H+) i okaże się, że obydwa wiązania H-O w wodzie to są wiązania koordynacyjne! Sytuacja jest więc dziwna, nieprawdaż? Bo kto mówi, że w cząsteczce wody mamy dwa wiązania koordynacyjne. No właśnie, nikt! Dlaczego? Woda ma przecież takie same właściwości – nie jest ważne, czy powstała z pierwiastków, rodników, czy z jonów z reakcji zobojętniania – powstaje ten sam związek i wiązania O-H są identyczne. Nieważne, czy wiązanie O-H powstało przez wniesienie po 1 elektronie przez każdy atom (sparowanie elektronów), czy też przez dostarczenie obu elektronów do wiązania przez jeden atom (bo drugi nie miał co wnosić), to powstaje to samo wiązania: O-H. Chlorek amonu możemy otrzymać w reakcji pomiędzy NH3 i HCl, ale możemy go też otrzymać w reakcji jonowego azotku sodu z kwasem solnym, np.

Azotek sodu Na3N jest zbudowany z jonów Na+ i N3-. Jeśli rozrysujemy sobie struktury elektronowe, to Okazuje się, że w tej reakcji anion azotkowy jest donorem wszystkich czterech par wiążących w jonie amonowym i wszystkie wiązania N-H w kationie amonowym są koordynacyjne! A jednak uczymy się, że tylko jedno z nich jest koordynacyjne. Skąd to wynika?
Wynika to stąd, że pierwiastki mają dwie tendencje do tworzenia związków chemicznych i osiągania konfiguracji helowca – jedna: uwspólnienie swoich niesparowanych elektronów walencyjnych – w tej sytuacji każdy atom wnosi po jednym elektronie do tworzącego się wiązania. Taką sytuację mamy np. w syntezie związków z pierwiastków. A druga przez uwspólnianie wolnych par elektronowych z atomem, który ma ich za mało by osiągnąć trwalszą konfigurację. W tej sytuacji to jeden z atomów wnosi oba elektrony do tworzącego się wiązania. Dopóki rozpatrujemy cząsteczkę, którą możemy otrzymać w reakcji chemicznej pomiędzy wolnymi pierwiastkami, dopóty nie rozpatrujemy powstających wiązań jako koordynacyjne, chociaż ten sam związek możemy otrzymać w reakcjach, gdzie donorem elektronów wiążących może być jeden atom. Dopiero rozpatrując indywiduum, którego budowy nie można wyjaśnić w oparciu o reakcję chemiczną pomiędzy atomami pierwiastków, to zakładamy, że któreś z wiązań jest koordynacyjne. Jonów H3O+ czy NH4+ nie otrzymamy w reakcji chemicznej pomiędzy atomami wolnych pierwiastków: H z O i H z N, ale nawet w takowej reakcji konieczne może być założenie obecności wiązania donorowo-akceptorowego, jak chociażby w przypadku cząsteczki SO2 jest. Wszystko co do tej pory przeczytaliśmy było po to, żeby pokazać, że wiązanie koordynacyjne to często tylko pojęcie formalne. Wiązania koordynacyjne możemy podzielić na „nieprawdziwe” i „prawdziwe”. W przypadku SO2 czy ma ono tylko charakter formalny – związane to jest z ograniczoną pojemnością powłoki walencyjnej, albo regułą oktetu. Przypadek dwutlenku siarki uświadamia nam, jak bardzo formalne może być pojęcie tego wiązania, gdy uwzględnimy rezonans elektronów. W niektórych przypadkach może to być wręcz niewłaściwe założenie, bo możemy mieć do czynienia z odstępstwami od reguły oktetu, a tak jest w przypadku i H3PO4 i H2SO4, gdzie edukacja szkolna zakłada istnienie wiązania koordynacyjnego, podczas gdy fosfor i siarka mają poszerzoną poziom walencyjny w stosunku do okresu drugiego. Z kolei za prawdziwe wiązanie koordynacyjne możemy uznać takie, gdy rozpatrywanej drobiny nie można otrzymać bezpośrednio z pierwiastków, a tworzenie drobiny związane jest z tendencją do osiąganie konfiguracji helowca przez wiążące się atomy, np. kation wodorowy (proton) H+ dąży do dubletu elektronowego – konfiguracji helu, co osiąga tworząc wiązanie koordynacyjne. Tak więc w jonie amonowym NH4+ czy hydroniowym H3O+ mamy po jednym prawdziwym wiązaniu koordynacyjnym – żadnego z tych jonów nie otrzymamy w reakcji pomiędzy atomami wolnych pierwiastków. Rozpatrzymy jeszcze jeden przypadek, mianowicie roztwarzanie Al(OH)3 w roztworze zasady:

Powstanie kompleksowy jon: anion tetrahydroksoglinianowy. Jak już możemy się spodziewać, jedno z wiązań jest wiązaniem koordynacyjnym, czyli kowalencyjnym. Co jakiś czas widzę pytania o charakter wiązań w tym jonie i pada też nieprawidłowa odpowiedź, że są to wiązania jonowe, bo przecież Al(OH)3 jest związkiem jonowym. Tymczasem wszystkie wiązania są równocenne – jeśli jedno jest koordynacyjne, czyli kowalencyjne, to pozostałe też są kowalencyjne. Problemy w tym przypadku sprawia fakt, że szkolne uproszczenia przyzwyczajają nas do myślenia, że wodorotlenki są jonowe z natury. Nic bardziej błędnego – nie mógłby powstać jon kompleksowy [Al(OH)4]–, gdyby którekolwiek wiązanie pomiędzy atomem glinu a grupą hydroksylową miało charakter jonowy. Gdyby tylko jedno wiązanie miało charakter kowalencyjny, to w roztworze byłby obecny jon Al(OH)2+, bo pozostałe jony OH– byłyby osobnymi drobinami obecnymi w roztworze, gdyż związki jonowe są w roztworach wodnych całkowicie zdysocjowane lub prawie całkowicie zdysocjowane (to temat na inną dyskusję).
Na koniec rozpatrzymy jeszcze jeden przypadek – cząsteczkę ozonu, czyli O3. Wspominam o tym celowo, ponieważ dostałem pytanie od pewnej studentki: czy w cząsteczce ozonu występuje wiązanie koordynacyjne? Jej zdaniem tak, ale ich wykładowca twierdzi, że nie. Ja na miejscu studentki zapytałbym wykładowcę, czy tlenek siarki(IV) zawiera wiązanie koordynacyjne – jeśli odpowiedziałby, że tak, to zaprzeczyłby sam sobie. Siarka i tlen jako swoje analogi mają identyczną strukturę walencyjną – struktura elektronowa ozonu i dwutlenku siarki jest identyczna – wystarczy centralny atom siarki zastąpić atomem tlenu i otrzymujemy cząsteczkę ozonu. Sami spójrzmy:

Wykładowca miał akurat rację, że jest to wiązanie pojedyncze, ale ze względu na ładunki elektryczne (związki takie nazywamy 1,2-dipolami) zakładamy, że jeden atom tlenu jest donorem tego wiązania. Trzeba tutaj jeszcze dodać, że wielu chemików uważa za wiązania koordynacyjne właśnie to, które tutaj nazwałem prawdziwym wiązaniem koordynacyjnym. Czy jest to słuszne podejście – oceńcie już sami.
Przebrnęliśmy. Wszystko to by wyjaśnić najbardziej palące kwestie, wskazać błędne myślenie o tym rodzaju wiązań. Mam jednak nadzieję, że choć częściowo to się udało. Ma też nadzieję, że przestaną być ze sobą mieszane różne i niezależne kryteria podziału wiązań kowalencyjnych.
Podsumujmy i zauważmy, jak źle są uczone wiązania chemiczne. Ile osób się zastanawia, czym się różni wiązanie kowalencyjne od koordynacyjnego. Albo czy wiązanie koordynacyjne liczy się jako σ itp., itd… A wystarczy tylko wytłumaczyć coś… Wiązanie koordynacyjne nie jest mitem. Ale mity są o nim opowiadane…