Poniższy tekst powstał ze względu na trwające prace nad drugą częścią podręcznika „Podstawy obliczeń chemicznych”. Ze względu na poruszone w nim zagadnienia ma bardziej rozbudowaną część opisową, niż część pierwsza, bazująca na podstawowych pojęciach. Tekst operuje zagadnieniami wprowadzonymi we wcześniejszych rozdziałach i ma charakter roboczy. Po redakcji całości materiału podręcznika ulegnie on skróceniu, a niektóre fragmenty tekstu mogą zostać przeniesione do innych rozdziałów. Artykuł ma charakter poglądowy i służy zapoznaniu czytelnika z charakterem podręcznika, oraz zebraniem uwag i sugestii.
Ten rozdział rozpoczniemy zupełnie inaczej, niż rozpoczyna się w innych podręcznikach. Rozpoczniemy od przyjrzenia się dwóm prostym, a jak się okaże – bardzo pomocnym doświadczeniom. Pierwsze z nich nawiązuje do rzeczy znanych już od szkoły podstawowej i do pierwszej części podręcznika. Drugie można wykonać np. na lekcji, a w uproszczonej wersji – także w domu. Dopiero po zapoznaniu się z nimi przejdziemy do omawiania bardziej podręcznikowych przypadków.
Doświadczenie 1:
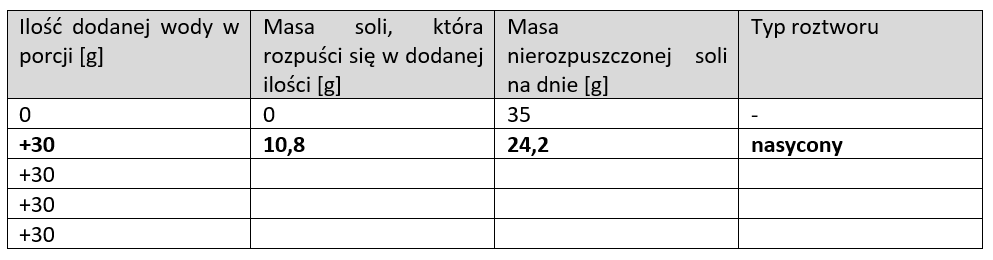
W zlewce lub szklance umieść 35 g chlorku sodu (soli kuchennej). Następnie dodaj 30 g (30 ml) wody o temperaturze pokojowej i mieszaj, aż osad soli wyraźnie przestanie się rozpuszczać. Dodaj kolejne 30 g wody i mieszaj, aż osad wyraźnie przestanie się rozpuszczać. Dodaj kolejne 30 g wody i mieszaj, aż osad przestanie się rozpuszczać. Potem dodaj kolejne 30 ml wody i mieszaj. Przerwij, gdy cała sól się rozpuści. Rozpuszczalność chlorku sodu w temperaturze pokojowej wynosi 36,00 g/100 g wody. Wypełnij tabelę:
Doświadczenie 2:
Wersja uproszczona: do dwóch dużych probówek (lub małych szklanek) wsyp w miarę możliwości równe masy wodorotlenku wapnia (można użyć budowlanego wapna suchogaszonego), najlepiej w przedziale 0,5 – 1,0 g. Następnie do jednej z nich równymi porcjami (np. po 2 ml) dodawaj octu spirytusowego. Po każdej porcji wstrząśnij probówką i obserwuj ilość osadu. Następnie dodaj porcję octu i powtórz czynności. Doświadczenie zakończ, gdy cały osad zaniknie. Powtórz czynności dodając identycznymi porcjami wodę do probówki drugiej. Doświadczenie zakończ, gdy do drugiej probówki wprowadzisz tę samą objętość płynu, co do pierwszej. Uwaga: techniczne wapno suchogaszone zawiera także niereagujące z kwasem octowym i praktycznie nierozpuszczalne zanieczyszczenia.
Wersja zaawansowana: w dwóch probówkach umieść po ok. 0,5 g suchego chlorku srebra AgCl. W przypadku jego braku można sporządzić świeży osad przez zmieszanie odpowiednich ilości NaCl i azotanu(V) srebra AgNO3, następnie ogrzać probówkę niemal do wrzenia jej zawartości (osad łatwiej opadnie na dno) i delikatnie usunąć jak najdokładniej roztwór znad osadu (np. pipetką Pasteura). Czynności związane z osadem chlorku srebra należy wykonywać z dala od światła słonecznego. Do suchego lub do mokrego osadu w pierwszej probówce należy porcjami po 1 ml dodawać r-r NH3 w wodzie o stężeniu 1 mol/dm3. Po dodaniu każdej porcji należy wstrząsnąć zawartością probówki i obserwować zachowanie się osadu. Gdy jego reakcja się zakończy, należy dodać kolejną porcję i ponownie obserwować osad. Gdy cały osad się roztworzy – dodaj jeszcze jedną porcję r-ru. Powtórz czynności dodając te same porcje wody do drugiej probówki do chwili, aż objętości dodanych płynów będą równe. Porównaj wygląd probówek. Jednomolowy roztwór amoniaku można otrzymać mieszając wodę z 25% wodą amoniakalną w stosunku objętościowym 1:12.
Nie uwzględniając przejściowego powstawania hydroksosoli, to równanie reakcji kwasu octowego z wodorotlenkiem wapnia wygląda następująco:

Jeśli przyjąć, że 1 ml octu spirytusowego zawiera 0,1013 g kwasu octowego to ten 1 ml roztwarza 62,5 mg wodorotlenku wapnia. Jak łatwo policzyć, póki na dnie pozostaje osad, to każda dodatkowa porcja octu daje w wyniku reakcji roztwór o tym samym stężeniu, co poprzednia porcja, póki nie wyczerpie się stały substrat – każdy ml octu roztwarza tę samą masę osadu i daje nową porcję roztworu o tym samym stężeniu.
Amoniak reaguje z osadem chlorku srebra wg równania stechiometrycznego:
![]()
Produktem jest dobrze rozpuszczalna sól kompleksowa. Jeden mililitr roztworu amoniaku o stężeniu 1 mol/dm3 zawiera 0,001 mol NH3, a zatem z równania reakcji wynika, że potencjalnie reaguje z 0,0005 mol (71,75 mg) AgCl. Oznacza to, że jeśli wykonać to doświadczenie wg podanych ilości, to osad powinien całkowicie się roztworzyć po dodaniu siódmej porcji (7 ml) roztworu. Póki osad się nie roztworzy całkowicie – otrzymujemy roztwór o tym samym stężeniu – każdy dodatkowy mililitr wody amoniakalnej nasyci się 71,75 mg osadu. Dodanie nadmiarowej porcji powoduje rozcieńczenie roztworu soli kompleksowej, bo wyczerpał się substrat. Jeśli do roztworu wprowadzić dodatkową porcję chlorku srebra – to zacznie ona się roztwarzać. Do doświadczeń nawiążemy jeszcze w tym rozdziale, na tę chwilę miejmy je w pamięci.
Dotychczas rozpatrywaliśmy wyłącznie reakcje w układach jednofazowych: w mieszaninach gazowych lub ciekłych – jest to łatwiej przyswajalny przypadek, dlatego od tego zwykle zaczyna się omawianie równowag. Układy niejednorodne mają swoją specyfikę i na nich się teraz skupimy. Uczniowie często pytają: czy ciała stałe uwzględniamy w stałej równowagi? Rozpatrzymy to razem, ale teraz już na typowo podręcznikowym przykładzie. Jeśli przez rurę wypełnioną koksem (węglem) C ogrzanym do temperatury żaru przepuszczać gorącą parę wodną, to powstaje mieszanina czadu CO i wodoru H2. Na wylocie z rury mieszanina poreakcyjna zawiera dodatkowo nieprzereagowaną parę wodną, którą łatwo oddzielić od pozostałych produktów przez obniżenie temperatury i skroplenie. Ostatecznym produktem jest więc gaz syntezowy – mieszanina CO i H2. Załóżmy teraz, że tę samą reakcję prowadzimy w reaktorze okresowym, tj. takim naczyniu, w którym nie ma ciągłego dostarczania przynajmniej jednego substratu i ciągłego odbierania przynajmniej jednego z produktów, tylko wszystkie substraty wprowadza się raz, następnie prowadzi się reakcję, a po odpowiednim czasie – odbiera się produkty lub wydziela się je z mieszaniny poreakcyjnej. Przykładem reaktora okresowego może być kolba lub probówka, w których przeprowadzamy jakieś reakcje chemiczne, choć nazwa reaktor w tym przypadku nie jest stosowana – raczej stosuje się ją do bardziej zaawansowanych naczyń, w których przeprowadza się reakcje chemiczne. Laboratoryjny reaktor okresowy z mieszadłem i płaszczem wodnym zapewniającym warunki izotermiczne (w tym przypadku 70 °C) przedstawia zdjęcie poniżej:

Zatem do reaktora okresowego wprowadzamy nadmiar grafitu (C), ogrzewamy układ do odpowiedniej temperatury i wprowadzamy parę wodną o tej samej temperaturze. W układzie rozpoczyna się odwracalna reakcja chemiczna opisana równaniem:

Reakcję prowadzimy w izochorycznym i izotermicznym układzie zamkniętym. Zamknięcie układu jest konieczne, bo o ile ciecze posiadają własną objętość, o tyle gazy nie posiadają jej, a zajmują całą dostępną przestrzeń – trzeba więc ją ograniczyć, bo w przeciwnym razie zaczną opuszczać układ dyfundując do otoczenia, a gazy obecne w otoczeniu do układu. Podczas reakcji rośnie ciśnienie w reaktorze, co jest związane ze wzrostem liczności cząsteczek reagentów gazowych w jej trakcie. Gdyby cała wodna przereagowała, to ciśnienie po reakcji byłoby dwukrotnością pierwotnego ciśnienia pary wodnej. Tak się jednak nie dzieje, bo układ osiąga stan równowagi wcześniej, zatem ciśnienie jest niższe od dwukrotności – tego się już domyślamy. W stanie równowagi układ zawiera stały grafit, parę wodną, tlenek węgla i wodór. Zauważmy, że układ zawiera dwie fazy: mieszaninę w gazowym stanie skupienia oraz ciało stałe. Składniki gazowe są zmieszane ze sobą na poziomie molekularnym, zajmują niemal całą objętość reaktora i mówimy o ich stężeniu w roztworze (mieszaninie gazowej), ale co w przypadku grafitu? Jak uwzględnić jego stężenie? Ten nie zajmuje całej objętości naczynia, jest skupiony w osobnej fazie i zajmuje tylko niewielką część objętości układu, taką, że przestrzeń dostępna dla gazów jest praktycznie równa objętości reaktora. W przeciwieństwie do nich nie możemy mówić o stężeniu grafitu w układzie, ale atomy węgla zebrane w fazie stałej mają jakieś stężenie: 1 dm3 grafitu ma masę 2150 g, co oznacza, że w 1 dm3 grafitu znajduje się 179 mol atomów węgla – czyli stężenie węgla w fazie stałej wynosi 179 mol/dm3. Zapiszmy stężeniową stałą równowagi dla tej reakcji uwzględniając wszystkie składniki:

Teraz zauważmy jedną rzecz. Wraz z postępem reakcji zmianie ulega masa i liczność wszystkich reagentów, rośnie lub maleje, nie ulega zmianie objętość układu (Vr). Stężenie reagentów gazowych się zmienia:

Ale co w przypadku grafitu? Zmienia się jego masa i objętość, ale nie ulega zmianie stężenie atomów węgla w sieci krystalicznej – podobnie jak gęstość nie zależy od ilości substancji, tak samo stężenie czy roztworu, czy czystej substancji nie zależy od jej ilości. Nie ma to znaczenia, czy kryształ grafitu waży 5 g, czy 20 kg – zawsze w 1 dm3 będzie tyle samo atomów węgla. Zatem jeśli w wyniku reakcji chemicznej zmieni się masa grafitu, to nie zmieni się jego stężenie w fazie stałej. Zatem i na początku, i w stanie równowagi stężenie tego składnika wynosi 179 mol/dm3:

W wyrażeniu na stałą równowagi mamy zatem dwie stałe: stałą równowagi oraz stężenie grafitu. Rozdzielmy więc stronami stałe od zmiennych:

Iloczyn dwóch stałych liczb także jest stałą liczbą – tę liczbę oznaczmy Kc’ i otrzymamy:

Czyli:

I tę nową stałą będącą iloczynem stałej równowagi i stężenia węgla w fazie stałej stosujemy w praktyce jako stałą równowagi tej reakcji. Często spotyka się dwa stwierdzenia: pierwsze, że w stałej równowagi nie uwzględniamy ciał stałych lub też, że stężenie ciał stałych wynosi 1 i dlatego ich nie zapisujemy. O ile pierwsze stwierdzenie nie jest całkowicie pozbawione sensu (w praktycznie używanej stałej ciała stałe się nie pojawiają), o tyle drugie jest nieprawdą. W przypadku omawianej przez nas reakcji wyglądało by to następująco:

Ale teraz jesteśmy świadomi, że nie wynosi 1, bo np. dla grafitu w temperaturze pokojowej wynosi 179, ale może być równe 1 tylko dla jakiejś konkretnej substancji chemicznej. W praktycznie używanej stałej równowagi stężenie ciał stałych już jest zawarte w jej wartości. Możemy powiedzieć, że jest ono tam ukryte. Zabieg taki znacznie ułatwia posługiwanie się stałymi równowagi, bez konieczności znajomości odpowiednich danych o gęstości ciała stałego w danych warunkach itp.
1) Czy podane wyprowadzenia są słuszne także dla następujących przypadków:
-reakcji prowadzonych w fazie gazowej, w której jeden z reagentów jest cieczą (np. tworzy mgłę w układzie reakcyjnym, lub ciecz stanowi fazę dolną, a gazowa górną)?
-reakcji prowadzonej w ciekłym roztworze, ale jeden z produktów jest cieczą praktycznie nierozpuszczalną w tym rozpuszczalniku i opuszcza środowisko reakcji tworząc emulsję.
2) Zastanów się, czy jest możliwe, że w przypadku odwracalnej reakcji opisanej równaniem:
![]()
Cały grafit ulegnie przereagowaniu?
Powyższe nasze rozważania są przykładem równowagi międzyfazowej. O ile dodatek lub ubytek któregoś reagenta będącego w gazowym stanie skupienia wpływa na jego stężenie, o tyle dodatek lub ubytek grafitu nie zmienia stężenia tego reagenta – położenie stanu równowagi nie zależy więc od wprowadzonej ilości grafitu do układu – zawsze przereaguje go jednakowa masa. Zwiększenie początkowej ilości tego substratu (nadmiaru, w naszym przypadku) nie przesunie położenia stanu równowagi. A co się stanie, gdy zmniejszymy ilość grafitu?
Rozpatrzmy razem następujący przypadek:
Przykład 1. w reaktorze o stałej pojemności 2 dm3 wykonujemy dwa doświadczenia w identycznych warunkach. W pierwszym wprowadzono 0,2 mol grafitu i 0,3 mol H2O. Następnie szczelnie zamknięto reaktor i ogrzano do temperatury 560 ℃. Gdy ciśnienie w kolbie przestało się zmieniać w czasie, to stwierdzono, że układ zawiera 0,05 mol grafitu i po 0,15 mol pary wodnej, tlenku węgla(II) i wodoru. W kolejnym doświadczeniu wprowadzono 0,1 mol grafitu i 0,3 mol H2O. Po szczelnym zamknięciu układ ogrzano do 560 ℃. Ile grafitu pozostało w układzie, gdy ciśnienie w reaktorze osiągnęło stałą wartość?
Doświadczenie wykonujemy w tych samych warunkach, ale zmniejszyliśmy ilość węgla. Jakie skutki to za sobą pociągnie? Temperatura i początkowe stężenie pary wodnej są w obu doświadczeniach identyczne, stała równowagi, wyrażona równaniem:

wynosi 0,075. Równowagowe ilości pary wodnej, tlenku węgla(II) oraz wodoru ponownie wyniosą 0,15 mol – bo warunki są identyczne. Żeby układ osiągnął stan równowagi, to w obu przypadkach musi przereagować 0,15 mol węgla. W pierwszym doświadczeniu układ zawierał potrzebną ilość węgla, ale w drugim – zabraknie 0,05 mol. Co to oznacza w praktyce? Że przereaguje cały grafit, zanim układ osiągnie stan równowagi. W układzie powstanie więc po 0,1 mol CO i H2, a zostanie 0,2 mol nieprzereagowanej pary wodnej. Zauważmy teraz na pozór paradoksalną rzecz – gdyby węgiel był w gazowym stanie skupienia, to jego ilość wpływałaby na położenie stanu równowagi, nie reagowałby całkowicie, a co więcej w obu przypadkach stan równowagi byłby inny. Ponieważ jednak stan równowagi nie zależy od jego ilości, to zawsze w danych warunkach musi go przereagować tyle samo, by stan równowagi został osiągnięty. Jest potrzebna pewna krytyczna ilość węgla, poniżej której równowaga nie zostanie osiągnięta ze względu na wyczerpanie się tego substratu. A zatem: jest możliwe całkowite przereagowanie substratu w reakcji równowagowej, w sytuacji, gdy reakcja przebiega chemiczna w układzie heterogenicznym (wielofazowym). Dodatek grafitu do naszego układu spowoduje, że para wodna zacznie reagować z nim do momentu, aż skład fazy gazowej wyrówna się ze składem otrzymanym w pierwszym doświadczeniu – wtedy dopiero układ osiąga stan równowagi. Dalszy dodatek grafitu nie spowoduje żadnych zmian w układzie. Gdybyśmy chcieli, aby w tych samych warunkach, czyli w tej samej temperaturze i przy tym samym ciśnieniu początkowym pary wodnej przereagowało więcej grafitu, to reakcje musimy prowadzić w większym naczyniu – wtedy układ będzie zawierał większą liczbę cząsteczek pary wodnej, która może przereagować z węglem, a więc więcej węgla przereaguje, nim układ osiągnie stan równowagi.
Teraz możemy wrócić do naszych doświadczeń opisanych we wstępie. Amoniak, chociaż jest w postaci ciekłego roztworu, zachowuje się analogicznie jak gazowy wodór w opisanym przypadku: jeśli chcemy, by roztworzyła się większa ilość chlorku srebra, to musimy dodać większą objętość rozpuszczalnika (a raczej: roztwarzalnika), czyli zwiększyć objętość układu (rysunek).

Można także zwiększyć stężenie amoniaku lub pary wodnej, ale rozpatrujemy sytuację, w której początkowe stężenie dla poszczególnych przypadków jest to samo, zarówno w przypadku reagenta ciekłego, jak i gazowego. Zauważmy, że nawet w przemianie fizycznej, czyli rozpuszczaniu NaCl w wodzie zachowanie się układu jest analogiczne, a nawet rozpuszczalnik ma stałe stężenie początkowe. We wszystkich przypadkach otrzymujemy ten sam typ równowagi pomiędzy dwoma fazami, a położenie stanu równowagi nie zależy od ilości początkowej składnika stałego. Woda nasyca się chlorkiem sodu, a stężenie tej substancji w nasyconym roztworze nie zależy od ilości chlorku sodu, woda amoniakalna nasyca się chlorkiem srebra (w postaci związku kompleksowego), a stężenie kompleksu w nasyconym roztworze także nie zależy od ilości chlorku srebra, a o przypadku z grafitem możemy powiedzieć, że układ nasyca się węglem (w postaci CO). Wprowadzona ilość takiego składnika nie wpływa na położenie stanu równowagi, a więc i na stałą równowagi, jak wcześniej ustaliliśmy. W przypadku nadmiaru czynnika powodującego przechodzenie fazy stałej do roztworu – nie osiągamy stanu równowagi, czyli układ jest nienasycony danym składnikiem. Zapamiętajmy tę analogię reakcji chemicznej do procesu fizycznego, jakim jest rozpuszczanie – bo rządzą nimi te same prawa. Gazy nie mają własnej objętości – zajmują całą dostępną przestrzeń, dlatego o ile rozpuszczanie chlorku sodu czy roztwarzanie chlorku srebra można wykonać w układzie otwartym, o tyle w przypadku gazów aby miały one identyczne stężenie – musimy prowadzić proces w układzie zamkniętym i dobrać odpowiednią wielkość reaktora.
Ciecze są praktycznie nieściśliwymi płynami – ich objętość praktycznie nie zależy od ciśnienia. Rozpuszczalność chlorku sodu zależy od temperatury. Zastanów się, czy gdyby ciekła woda była płynem ściśliwym (jej objętość zmieniała by się wraz z ciśnieniem, jak u gazów), to czy rozpuszczalność chlorku sodu w wodzie zależałaby tylko od temperatury, czy też od temperatury i ciśnienia?
Aby zakończyć wywody rozpatrzymy jeszcze jedną, na pozór absurdalną sytuację. Syntezę jodowodoru z pierwiastków, ale w dwóch wariantach: w fazie gazowej (w układzie jednorodnym), oraz w układzie dwufazowym, z udziałem stałego jodu. W pierwszym przypadku sytuacja wygląda następująco:

A w drugim:

Stałe te nie są sobie równe! Przyjrzyjmy się, jak zachowuje się stały jod w warunkach reakcji. Czy pamiętacie doświadczenie z sublimacją i resublimacją kwasu salicylowego lub jodu? Jeśli tak, to dobrze, bo nawiążemy do tego. Jod także sublimuje, stąd kryształy jodu mają zapach podobny do chloru. Prężność par jodu rośnie wraz z temperaturą, co jest logiczne. Jeśli do pojemnika próżniowego wrzucimy kryształek jodu, to kryształek będzie sublimował tak długo, aż układ osiągnie równowagę, co zobaczymy jako słabsze lub mocniejsze nasycenie fioletową barwą (zależnie od wielkości naczynia i temperatury utrzymywanej w układzie, patrz obrazek).
")
Jeśli do tego naczynia wprowadzić jeszcze wodór, to zaczyna on reagować z parą jodu. Powoduje to spadek ciśnienia pary jodu, czyli otrzymamy parę nienasyconą. Stały jod zacznie więc sublimować do momentu, by odtworzyć stan równowagi ciało stałe-para – obecność stałego jodu powoduje, że nie zmienia się stężenie par jodu w układzie. Barwa układu pozostanie niezmienna, nawet jeśli zwiększymy lub zmniejszymy ciśnienie wodoru – a pamiętamy, że obecność innych lotnych składników nie wpływa na prężność pary nasyconej. Gdybyśmy jednak wrzucili do ampuły zbyt małą ilość krystalicznego jodu, by osiągnąć prężność pary nasyconej, to cały jod wysublimuje i wtedy w trakcie reakcji stężenie jodu się zmienia, bo nie ma możliwości, by ubytek uzupełnić poprzez sublimację odpowiedniej ilości jodu. Podobnie chlorek srebra nieznacznie rozpuszcza się w wodzie i póki mamy osad na dnie – stężenie chlorku srebra w roztworze jest stałe. Poruszone przypadki są procesami etapowymi. Spróbujmy się temu przyjrzeć rozpatrując równowagę poszczególnych etapów. Najpierw dla sublimacji jodu, a potem dla syntezy jodowodoru:

Dodajmy do siebie powyższe równania elementarne i otrzymamy znane nam już równanie sumaryczne (3):

A zatem:

Po skróceniu:

A po rozdzieleniu stałych stronami:

Mamy więc matematyczne potwierdzenie, że obecność reagenta znajdującego się w poza fazą zachodzenia reakcji nie wpływa na położenie stanu równowagi. Tak jak jod częściowo sublimuje, tak chlorek srebra czy wodorotlenek wapnia – nieznacznie się rozpuszczają w wodzie, a o położeniu stanu równowagi reakcji roztwarzania tych związków decyduje nie prężność pary nasyconej, lecz rozpuszczalność, czyli stężenie roztworu nasyconego.
Zawsze, gdy układ zawiera jakiś reagent skupiony poza fazą zachodzenia reakcji (roztworem) to osiąga on stan równowagi, gdy stężenie tego składnika w roztworze reakcyjnym może osiągnąć nasycenie. Przykłady tego typu układów to układ dwóch cieczy o ograniczonej rozpuszczalności (emulsja lub ciecze rozdzielone na warstwę górną i dolną), cieczy w gazie (mgła lub ciecz znajduje się na dnie naczynia), ciała stałego w cieczy (zawiesina lub osad), czy ciała stałego w gazie (ciało stałe na dnie naczynia lub rozpylone w gazie). Reakcja nie musi zachodzić tylko po rozpuszczeniu się lub odparowaniu/wysublimowaniu składnika do roztworu reakcyjnego – może zachodzić także na granicy (styku) faz – ale mimo tego nie ma to wpływu na położenie stanu równowagi. Reakcja na granicy faz jest to tylko równoległą ścieżka prowadząca układ do tego samego stanu. Gdyby nie reakcja na granicy faz, to to niektóre reakcje biegłyby bardzo powoli, np. otrzymywanie gazu syntezowego w reakcji pary wodnej z grafitem (lub w skali przemysłowej: z koksem) trwałoby bardzo długo, gdyż węgiel jest substancją niemal zupełnie nielotną do bardzo wysokich temperatur, a obecność lotnych składników nie podnosi lotności tego pierwiastka.