
W tym opracowaniu zebrałem najbardziej palące kwestie, które towarzyszą abiturientom w przededniu matury. Są to kwestie zazwyczaj kłopotliwe i łatwo wywołujące spory wśród maturzystów. Zestaw ten obejmuje zagadnienia, o które pytania pojawiają się regularnie, co oznacza, ze sprawiają największe kłopoty.
Polecam także nasz kanał w serwisie YT, na którym można obejrzeć wiele filmów dotyczących wielu zagadnień oraz do przejrzenia spisu treści czasopisma „Antocyjan”, w którym także publikujemy materiały dla osób szykujących się do egzaminu maturalnego.
1. Stopnie utlenienia
1) Stopień utlenienia wolnego pierwiastka zawsze wynosi 0 – nie ma tutaj znaczenia, w jakim stanie skupienia czy odmianie alotropowej dany pierwiastek występuje. Zerowy stopień utleniania mają zarówno ditlen O2, jak i tritlen O3, czyli ozon. Podobnie nie ma znaczenia, czy mówimy o graficie, czy diamencie, czy może fulerenie – wszystkie mają zerowy stopień utlenienia. Kłopotliwy dla maturzystów często jest ozon.
Definicja stopnia utlenienia jest jedna – nie ma potrzeby sugerowania się różnymi regułkami, o ograniczonym zastosowaniu i które nieraz są źródłem problemów, bo wystarczy wiedzieć czym jest dane pojęcie. Problematyczne są związki organiczne dla licealistów – nie bez powodu, bo nie znając definicji jakiegoś pojęcia trudno się nim posługiwać, gdy zabraknie jakiejś kolejnej regułki.
2) Stopień utlenienia atomu azotu w grupie nitrowej określamy jako III, a nie V, jak często automatycznie sądzimy biorąc pod uwagę, że o nitrowaniu myślimy w kategorii używania kwasu azotowego(V). Nitrowanie z użyciem HNO3 jest reakcją redoks – azotowi przypisujemy redukcję, a atomowi węgla, do którego przyłącza się grupa nitrowa – utlenienie, podobnie jak jak wprowadzając grupę OH zamiast atomu wodoru w cząsteczce metanu otrzymujemy metanol, czyli substancję, w której węgiel uległ utlenieniu, tak samo zastępując wiązanie C-H wiązaniem C-N podnosimy stopień utlenienia węgla – coś też musiało się zredukować. Związki nitrowe można otrzymać także w reakcji substytucji nukleofilowej z azotanów(III):

A nikt nie będzie się kłócił o to, że w jonie azotanowym(III) atom azotu ma V stopień utlenienia. Przez błędne założenie V stopnia utlenienia atomu azotu wiele osób nie potrafi potem zbilansować równania redukcji nitrobenzenu do aniliny, bo nie można dobrać współczynników stechiometrycznych.
Polecam: stopnie utlenienia w związkach organicznych
2. Bilansowanie metodą jonowo-elektronową
Podczas bilansowania równań reakcji redoks metodą jonowo-elektronową nie używa się stopni utlenienia – tam bilansuje się używając cząsteczek wody oraz jonów OH–, H+ (zależnie od środowiska), a bilans ładunku wyrównuje się brakującymi elektronami. Jony lub cząsteczki generowane w jednym równaniu połówkowym muszą być użyte w drugim równaniu połówkowym, podobnie jak elektrony. Nie można w jednym równaniu generować środowiska kwasowego, a w drugim zasadowego równocześnie, „bo to się potem zobojętni”. Większość uczniów ma wpojoną błędną metodę dobierania bilansu określając najpierw stopnie utlenienia, których używa się w bilansowaniu metodą elektronową. Nieraz są problemy z uzgodnieniem równania, co wynika z trudności w ustaleniu stopni utlenienia, które są w tej metodzie zbędne. Trudność ta dotyczy też wspomnianego wyżej ozonu w roli utleniacza, który przechodzi w m.i. w ditlen. A przecież wystarczy zapisać równanie połówkowe redukcji:

i nie było potrzeby wnikać w stopnie utlenienia, bo to nie ta metoda bilansowania.
Dla zainteresowanych: Bilans jonowo-elektronowy i stopnie utlenienia
3. Protonowanie a uwodornienie
Czasem słyszy się błędne opinie, że biologiczna redukcja polega na protonowaniu – nie, gdyż wprowadzenie kationu wodoru nie powoduje zmiany stopnia utlenienia. W reakcjach zachodzących w układach biologicznych biorą udział protony, ale równocześnie do redukcji potrzebne są elektrony. Jeden kation wodoru i jeden elektron odpowiada atomowi wodoru, a 2H+ + 2e- – cząsteczce wodoru. Redukcja polega na uwodornieniu, a nie na protonowaniu. Protonowanie nie jest reakcją redoks, a kwasowo-zasadową. Układy biologiczne nie są przystosowane do używania wolnego wodoru, stąd redukcja biegnie wg innego mechanizmu, ale efekt jest ten sam.
Dla zainteresowanych: protonowanie, czy uwodornienie?
4. Rozpuszczalność i nierozpuszczalność
często możemy usłyszeć, że jakaś substancja jest nierozpuszczalna, lub jest osadem. Jest to błędne myślenie z dwóch powodów – nic samo z siebie nie jest osadem, jako osad może zostać w jakichś warunkach otrzymane (zwykle w reakcjach wykonywanych w probówkach). Można strącić osad gipsu (CaSO4*2H2O), a można otrzymać piękne kryształy gipsu. Druga sprawa to kwestia rozpuszczalności – substancje dzielimy na dobrze rozpuszczalne, słabo (trudno) rozpuszczalne i bardzo słabo rozpuszczalne. Te ostatnie zwiemy PRAKTYCZNIE nierozpuszczalnymi, a nie nierozpuszczalnymi, bo takowych związków albo nie ma wcale, albo są to niezwykle rzadkie przypadki. Nazywanie substancji nierozpuszczalnymi jest pospolitym błędem. Każdy strącany osad jest w jakimś stopniu rozpuszczalny i nigdy strącanie nie zachodzi w 100% – bo jakaś część związku pozostanie w roztworze. Co najwyżej co najwyżej można otrzymać osad z wydajnością niemal 100% (praktycznie 100%). Substancje te praktycznie nierozpuszczalnymi, bo ze względu na bardzo niskie stężenie nie nadają się do przeprowadzania większości doświadczeń. Aby w temperaturze pokojowej rozpuścić 1 g CaCO3 potrzeba ponad 70 dm3 wody. „Woda” nad „nierozpuszczalnym” Mg(OH)2 ma odczyn zasadowy i barwi fenoloftaleinę na malinowo.
5. Zasady zapisu jonowego
- Często słyszymy: to nie dysocjuje, bo to osad. Stwierdzenie to nie jest poprawne. Jeśli w zapisie jonowym nie rozpisujemy czegoś na jony, to są ku temu 2 powody: powstaje słaby elektrolit (zdysocjowany w niewielkim stopniu, np. w reakcji wypierania kwasu octowego) lub dany związek opuszcza środowisko reakcji. W tym drugim przypadku oznacza to, że albo w reakcji strąca się osad, powstaje emulsja lub układ rozdziela się na dwie fazy ciekłe, lub też, że wydziela się gaz. Czy amoniak nie dysocjuje, bo jest gazem? Jeśli rozpatrujemy gaz, to nie dysocjuje, ale posiada zdolność do dysocjacji w wodzie – jeśli rozpatrujemy roztwór amoniaku, to możemy mówić o jego dysocjacji. Podobnie osady jako faza stała też nie są zdysocjowane, ale ta część, która przejdzie do roztworu lub nie ulegnie strąceniu (bo każdy osad jest w jakimś stopniu rozpuszczalny) – dysocjuje, jeśli spełnia ku temu warunki, np. jest to związek jonowy. W wodzie nad osadem CaCO3 są jony wapnia jak i węglanowe, tylko ich stężenie jest niskie, ze względu na rozpuszczalność tego związku. Ta woda jest w istocie roztworem.
- Ca(OH)2 w zapisie jonowym: to problematyczny przypadek z powodu nie do końca jasnego przekazu o zasadach zapisu jonowego. Jeśli rozpatrujemy roztwór Ca(OH)2 (wodę wapienną) – to zapisujemy go jonowo, bo jest to mocny elektrolit. Jeśli natomiast roztwarzamy stały Ca(OH)2, lub strącamy go jako osad, to zapisujemy go w postaci wzoru sumarycznego.
Tak uwzględniamy fakt, że albo początkowo związek ten nie był składnikiem roztworu, albo że ten roztwór częściowo opuszcza – bo rozpuszczalność Ca(OH)2 nie jest zbyt niska. W odpowiednich warunkach możemy strącić NaCl – wtedy w zapisie jonowym także posłużymy się wzorem sumarycznym, czyli NaCl. W procesie Solvaya strąca się NaHCO3 (soda oczyszczona) i w zapisie jonowym uwzględniamy ten fakt zapisując wzór sumaryczny, a przecież nie jest to związek trudno rozpuszczalny – po prostu w warunkach prowadzenia procesu się wytrąca.
6. Tabela rozpuszczalności i jej zastosowanie
Tabela rozpuszczalności to szczególne narzędzie służące do określania kierunku reakcji w wodnych roztworach. Dotyczy to reakcji typu sól+sól lub sól+zasada. I tylko do tego. Reakcja zajdzie, jeśli powstaje produkt trudno rozpuszczalny lub praktycznie nierozpuszczalny. Nie możemy jednak na podstawie tabeli rozpuszczalności określać kierunku reakcji innych, jak sól+kwas. Często w takich przypadkach pokutuje przekonanie, że jeśli powstaje osad, to można przeprowadzić reakcję w kierunku przeciwnym, niż wynika z mocy kwasów. Jest to bardzo błędne myślenie, bo aby móc stwierdzić, że słaby kwas wyprze mocny kwas, bo powstaje osad – potrzebne są konkretne dane liczbowe, wyznaczone doświadczalnie: stałe równowagi. Nie możemy posiłkować się tabelą rozpuszczalności, by stwierdzić, że przepuszczając dwutlenek węgla przez wodny roztwór np. CaCl2 czy Ca(NO3)2 reakcja zajdzie, bo otrzymamy osad CaCO3. Związek ten, choć praktycznie nierozpuszczalny, to roztwarza się nawet w słabym kwasie octowym, a przecież w reakcji o której powstaje poza osadem mocny kwas (HCl lub NHO3). Nie można strącić osadu CaCO3 ( i innych węglanów) tą metodą. Za to w przypadku przepuszczania gazowego H2S przez roztwory mocnych kwasów z kilkoma metalami (Pb, Cu, Ag, Hg) można otrzymać osad siarczku, ale nie jest to metoda ilościowa – reakcja zajdzie tylko częściowo. Wynika to stąd, że każdy związek trudno i bardzo trudno rozpuszczalny wymaga pewnego krytycznego stężenia kwasu, aby doszło do wyparcia słabego kwasu. Póki mocny kwas nie osiągnie tego stężenia w trakcie zachodzenia reakcji, osad będzie powstawał. Dlatego też możliwe jest otrzymanie osadu w przypadku przepuszczania H2S przez wodny roztwór CuSO4 czy Cu(NO3)2:

Strzałki równowagi wskazują, że jest to reakcja równowagowa – dodatek mocnego kwasu może spowodować przesunięcie tego stanu w lewo i całkowicie roztworzyć osad.
Na podstawie samej tabeli rozpuszczalności nie możemy mieć pewności, że reakcja sól+kwas zajdzie w kierunku przeciwnym do wynikającego z mocy kwasów. Dlatego też GDY POLECENIE WYRAŹNIE NA TO NIE WSKAZUJE, to mając do dyspozycji jedynie tabelę rozpuszczalności osady należy otrzymywać w reakcji sól+sól lub sól+wodorotlenek. Inna sytuacja to przypisany zestaw odczynników do wyboru, w których jedyną możliwości jest otrzymać osad w reakcji sól+kwas. Praktycznie nierozpuszczalne są liczne wodorotlenki, ale osadów wodorotlenków nie otrzymuje się poprzez hydrolizę soli. Hydroliza zachodzi w zbyt mały stopniu, i zazwyczaj nie towarzyszy jej powstanie osadu. Stan równowagi hydrolizy zazwyczaj jest na tyle przesunięty w lewo, że nie przekracza to rozpuszczalności wodorotlenku, dlatego np. CuSO4 tworzy piękne niebieskie roztwory, zamiast niebieskiej galarety w wodzie. Jeśli strąca się wodorotlenek (lub hydroksosól), to albo jest to zmętnienie, albo osadu jest niewiele (do ustalenia się równowagi, patrz przypadek z H2S). Tabela rozpuszczalności nie pozwala nam przewidzieć, czy podczas hydrolizy powstanie osad wodorotlenku, dlatego nie mając do dyspozycji odpowiednich danych, osady wodorotlenków strącamy w reakcji z dobrze rozpuszczalnym wodorotlenkiem.
Często jeden praktycznie nierozpuszczalny związek można przeprowadzić w inny praktycznie nierozpuszczalny związek. Warunkiem zajścia takiej reakcji jest przejście osadu bardziej rozpuszczalnego w mniej rozpuszczalny. Np. biały AgCl w obecności dobrze rozpuszczalnego jodku (np. KI) przechodzi w żółty AgI, ale nigdy na odwrót. AgI jest osadem o mniejszej rozpuszczalności, więc zajdzie reakcja AgCl + KI. Tabela rozpuszczalności nie pozwala nam jednak przewidzieć, kiedy możliwe jest przeprowadzenie jednego osadu w inny osad, a skupia się na reakcjach, jakie zachodzą w roztworach dobrze rozpuszczalnych związków po ich zmieszaniu ze sobą – osad może być tylko produktem. Tylko tyle można odczytać z tabeli rozpuszczalności.
7. Hydroliza soli
1) Roztwory wodne wielu soli nie mają obojętnego odczynu ze względu na ich hydrolizę. Jednak o hydrolizie krąży wiele niezgodnych z rzeczywistością wyobrażeń. Najpierw musimy sobie zdawać sprawę z tego, że hydroliza większości soli zachodzi w nieznacznym stopniu, np. pH roztworu octanu sodu o stężeniu 0,1 mol/dm3 w temperaturze pokojowej wynosi niespełna 9, a gdyby cały uległ hydrolizie zasadowej – wynosiło by 13. Jeśli pH wynosi 9, to oznacza to, że w roztworze o takim stężeniu hydrolizie ulega mniej niż 1 na 10 000 jonów octanowych obecnych w roztworze. Pomiędzy zobojętnianiem a hydrolizą istnieje stan równowagi. Często panuje nieprawdziwe przekonanie, że w hydrolizie NaHCO3 wydziela się CO2, a w hydrolizie NH4Cl wydziela się gazowy NH3 o gryzącym zapachu. Nie jest to prawdą – czy ktokolwiek widział, by wydzielał się CO2 w kontakcie sody oxczyszczonej z wodą? Albo czy czuć zapach amoniaku podczas rozsypywania na wilgotną glebę nawozy amonowe, jak (NH4)2SO4, czy NH4NO3? W przypadku hydrolizy kationu, jak wspomniałem wcześniej, przeważnie nie powstaje osad wodorotlenku w hydrolizie soli słabej zasady z mocnym kwasem, odczyn takich roztworów jest lekko kwasowy, a osadu albo nie ma wcale, albo powstaje go niewiele (np. jako zmętnienie). Nieco inaczej sytuacja wygląda w przypadku soli słabych zasad ze słabymi kwasami – tutaj hydrolizie ulega zarówno kation, jak i anion. Hydroliza anionu na tyle podnosi pH (w okolice odczynu obojętnego), że jest możliwe strącenie osadu. I tak w roztworze CuSO4 tworzy piękne i klarowne roztwory, ale już podczas rozpuszczania octanu miedzi w wodzie powstaje nieznaczna ilość osadu będącego głównie hydroksosolą z domieszką Cu(OH)2. Ten sam kation ulega hydrolizie w większym stopniu w soli z kwasem słabym, niż w soli z kwasem mocnym – zapach amoniaku pojawia się po rozpuszczenia NH4HCO3 w wodzie – czy wiesz już, dlaczego?
Stopień hydrolizy wzrasta wraz z rozcieńczanie roztworu i czasem osad pojawia się podczas rozcieńczania roztworu.
2) Kolejnym problemem jest często błędny zapis równań hydrolizy wynoszony ze szkoły. Jeśli hydrolizie ulega sól kwasu wieloprotonowego (H3PO4 czy H2S), to tak jak wolny kwas dysocjuje etapami, a każdy etap zachodzi w innym i coraz mniejszym stopniu, tak hydroliza jest procesem etapowym. Bo dysocjacja i hydroliza są ze sobą ściśle związanymi wielkościami, a czynnikiem je wiążącym jest iloczyn jonowy wody (Kw). I tak jak kwas ma więcej niż jedną stałą dysocjacji (Ka), tak anion kwasu ma więcej niż jedną stałą hydrolizy. Obecnie apeluje się do nauczycieli o zaprzestanie stosowania zupełnie błędnego i nielogicznego zapisu hydrolizy w jednym równaniu sumarycznym, np.:

Czy też często spotykane:

Prawidłowo hydrolizę zapisujemy etapami, np.:

Aby udowodnić odczyn roztworu soli ulegającej hydrolizie wystarczy zapisać pierwszy jej etap – bo ma decydujący wpływ na pH roztworu, podobnie jak pierwszy etap dysocjacji niemal zupełnie odpowiada za pH roztworu kwasu. Obecnie apeluje się do nauczycieli, o zaprzestanie stosowania tego błędnego zapisu.
3) Często panuje przekonanie, że związki praktycznie nierozpuszczalne nie ulegają hydrolizie, ani dysocjacji. Jest to błędne, ponieważ jakaś część związku się rozpuści, a to to się rozpuści – podlega dokładnie tym samym prawom, co roztwory związków dobrze rozpuszczalnych. W temperaturze pokojowej nasycony roztwór CaCO3 (prawie woda) ma stężenie 0,0014% i 0,00014 mol/dm3 i ma takie samo pH, co roztwór Na2CO3 o tym samym stężeniu molowym. Bo anion węglanowy ulega w ty roztworach hydrolizie w takim samym stopniu. Nie hydrolizuje sam osad, tylko to, co przeszło do roztworu (ta część osadu, która się nie strąciła ze względu na częściową rozpuszczalność strącanej substancji). Nie hydrolizuje stały NaHCO3 (np. strącony w procesie Solvaya), tylko ten, który jest w roztworze.
W przypadku azotanu srebra często można spotkać nieprawidłowe stwierdzenie, że związek ten hydrolizuje do Ag2O, ze względu nietrwałość AgOH. Musimy jednak pamiętać, że nietrwały jest osad AgOH – strącony wodorotlenek srebra szybko przechodzi w trwalszy Ag2O. Jednakże pomiędzy reakcją strąceniową, a hydrolizą soli jest różnica. AgNO3 nie hydrolizuje do Ag2O, bo nie wytrąca się w roztworze tej osad, który przechodziłby w Ag2O.
Zainteresowanym polecam: Czy osady dysocjują?

4) Hydroliza mydeł. Postanowiłem dodać ten punkt, bo o hydrolizie mydeł często można spotkać herezje, z których wynika, że mydło parzy. Mydłami nazywamy sole wyższych kwasów karboksylowych, czyli tłuszczowych. Sole litowców są dobrze rozpuszczalne w wodzie, natomiast większość mydeł jest w wodzie praktycznie nierozpuszczalna. Jako środki myjące używane są mydła sodowe i potasowe, natomiast inne mydła mają zastosowania specjalne, np. wiele mydeł ma dobre właściwości smarne. Kwasy tłuszczowe są praktycznie nierozpuszczalne w wodzie oraz są kwasami słabymi. Z tego drugiego powodu sole kwasów tłuszczowych ulegają hydrolizie anionowej. Można jednak spotkać nieprawdziwe informacje o mydłach, przede wszystkim to, że w hydrolizie mydeł strąca się osad kwasów tłuszczowych, bo są praktycznie nierozpuszczalne w wodzie, oraz to, że ich hydroliza jest nieodwracalna, właśnie ze względu na wytrącanie się kwasu tłuszczowego:

Jednakże jest to zapis zupełnie błędny. Wynika z niego, że po rozpuszczeniu mydła w wodzie otrzymujemy zasadę sodową/potasową i osad kwasu tłuszczowego. Jaki sens miałoby w takim razie stosowanie mydła, skoro ono nie działa, bo działa zasada. Prościej byłoby rozpuścić trochę KOH lub NaOH w wodzie i tym umyć ręce. Miałoby to jednak fatalne skutki. Wyjaśniłem wcześniej, że hydroliza zachodzi zazwyczaj w tak małym stopniu, że albo osadów nie ma wcale, albo jest ich niewielka ilość. Tak więc i w tym przypadku hydroliza powoduje, że odczyn roztworu mydła jest lekko zasadowy (pomijając szare mydło, czyli czyste mydło, to handlowe mydła w kostce zawierają dodatki, które jeszcze dodatkowo obniżają pH r-ru mydła bo bardziej zbliżonego do pH skóry). Za właściwości myjące mydeł odpowiadają aniony karboksylanowe, które mają czynność powierzchniową. Zapis powyżej sugeruje, że ich w roztworze nie ma, bo przecież wszystkie uległy hydrolizie, wtedy za myjące właściwości musiały by odpowiadać wyłącznie jony OH– – co nie byłoby szczęśliwym dla nas rozwiązaniem, a dwa, mydło byłoby niepotrzebne, bo wystarczyło by rozpuścić w wodzie trochę KOH czy NaOH. W roztworze mydła dominują jony karboksylanowe. Z związku z tym w kontakcie z wodą twardą strącają się mydła wapniowe i magnezowe. Gdyby hydroliza zachodziła nieodwracalnie, to osadem w twardej wodzie byłby kwas tłuszczowy i dodatkowo wodorotlenki, które powstałyby w reakcji z wydzieloną zasadą, np. Mg(OH)2 mógłby się też wytrącać. Dlatego zapominamy o błędnym zapisie i opiniach o hydrolizie mydeł.

Oczywiście o ile polecenie tego jasno nie określa, to nie stracimy punktów za użycie jednej strzałki.
8. Odczyn a pH
Odczyn służy tylko do określania, której formy rozpuszczalnika w roztworze jest więcej: kationowej (kwasowej), czy anionowej (zasadowej). Jeśli jest ich równa ilość, to odczyn jest obojętny. Z kolei skala pH służy do wyrażania stężenia – jest miarą kwasowości i jest znacznie precyzyjniejszym pojęciem, niż odczyn. Odczyn może być tylko kwasowy, zasadowy, lub obojętny, podczas gdy pH ma wymiar liczbowy. Czasem spotyka się błędnie użycie słowa odczyn, jako synonimu pH, lub mówi się o odczynie pH. Zapamiętaj, że nie mówi się, że odczyn wynosi pH…, tylko odczyn jest kwasowy/zasadowy/obojętny, a pH wynosi konkretną liczbę.
9. Zobojętnianie i odczyn obojętny
Te pojęcia często są ze sobą mylone. Zobojętnić, a doprowadzić do odczynu obojętnego nie oznacza to samo. Zobojętnienia jest metodą otrzymywania soli, podczas gdy doprowadzanie do odczynu obojętnego oznacza w przypadku wodnych układów doprowadzenie do pH=7. W zobojętnianiu otrzymujemy sól obojętną, to jest sól, która nie zawiera nieprzereagowanych kwasowych protonów lub grup wodorotlenowych. Czyli zobojętniając np. kwas siarkowy (dwuprotonowy) otrzymujemy sól obojętną, czyli np. K2SO4, a nie KHSO4, oraz np. CaCl2, zamiast Ca(OH)Cl. Oznacza to więc taki stechiometryczny stosunek substratów do siebie, by powstała taka sól, zwana obojętną. Jeśli reakcję zobojętniania przeprowadzać w środowisku wodnym, to często odczyn nie jest obojętny mimo stechiometrycznego stosunku, ze względu na hydrolizę powstałej soli. Np. w przypadku zobojętniania kwasu octowego mocną zasadą zobojętnienie otrzymujemy przy pH niespełna 9 – bo takie jest pH roztworu czystego octanu sodu czy potasu. Dla zobojętniania amoniaku mocnym kwasem, np. solnym – oznacza to pH ok. 5 – bo takie pH ma roztwór czystego NH4Cl. Zapamiętaj też, że odczyn ma nie sama sól, tylko jej roztwór w wodzie.
10. Roztwory buforowe
Częstym problemem jest zapisanie równań reakcji składników buforu z kwasem, lub zasadą. Zapamiętaj, że dodając mocnego kwasu do roztworu buforowego w pierwszej kolejności reakcji ulega silniejsza zasada (bo chętniej przyjmuje proton), a dodając mocnej zasady – w pierwszej kolejności reaguje mocniejszy kwas – łatwiej pozbywa się protonu. W pospolitym buforze fosforanowym (roztworze zawierającym KH2PO4 i K2HPO4 w różnych stosunkach) mocniejszy kwas stanowi anion H2PO4–, a mocniejszą zasadę – anion HPO42-. Dopiero po wyczerpaniu się jednego z nich – zaczyna zachodzić reakcja z drugim jonem. Dlatego dodając kwasu do buforu fosforanowego zachodzi reakcja:

A dodając zasady:

Czy potrafisz napisać, jakie reakcje zajdą po wprowadzeniu kwasu lub zasady do buforu węglanowego, to jest roztworze zawierającym NaHCO3 i Na2CO3? Czy potrafisz wyjaśnić, dlaczego roztwór NH4HCO3 ma właściwości buforujące?
11. Stała równowagi
1) Jednostka. Dość pospolitym stwierdzeniem jest, że stała równowagi ma jakąś jednostkę, i są podręczniki, w których można tę pomyłkę spotkać. Stała równowagi jest wielkością bezwymiarową i niezależną od systemu jednostek. Jest wielkością logarytmowalną – nie można logarytmować jednostek, a jedynie liczby. Gdyby stała równowagi miała jednostkę, nie byłoby ani logarytmowalna (nie byłoby wartości pK), a ponadto przy jej wartości trzeba by dodatkowo podać o jaką jednostkę chodzi. Bo przecież nikt nas nie zmusza do wyrażania stężenia w mol/dm3 – można używać innych jednostek i zależnie od nich stała miałaby inną wartość, co utrudniło by wszystkim życie. Gdyby stała równowagi miała jednostkę, to i wartość pK ją miałaby. Aby otrzymać wielkość, która nie zależy od stałej równowagi wykonano bajecznie prostą sztuczkę – uznano, że wszystkie wartości, które podstawiamy do stałej równowagi, są podzielone przez stężenie standardowe (1, gdy jednostką stężenia jest mol/dm3, a w przypadku stałej równowagi wyrażonej przez ciśnienia – przez 1000 hPa lub w przypadku używania innych jednostek są to wielkości im odpowiadające, np. jeśli są nimi mmHg to dzielimy przez o 760 mmHg.
2) Stała równowagi i ciała stałe. Do stałej równowagi wstawiamy wszystkie reagenty, które biorą udział w reakcji. Jednakże jeśli któryś reagent występuje w innej fazie, tzn. nie znajduje się w fazie, w której zachodzi reakcja, to stężenie takiego reagenta nie zmienia się – stężenie takich reagentów nie zmienia się w trakcie zachodzenia procesu, bo nie są rozpuszczone. Zmienia się ich masa/objętość, ale nie stężenie. Stężenie np. cząsteczek sacharozy w krysztale sacharozy wynosi 4,65 mol/dm3 – nie ma znaczenia, czy jest to mały kryształek, czy duży – w trakcie procesu zmieni się jego wielkość – ale stężenie nadal będzie wynosić 4,65 mol/dm3. Dla przykładu rozpatrzmy reakcję daną równaniem:

Stężeniowa stała równowagi przyjmuje postać:

Jednakże węgiel stanowi osobną fazę – jest ciałem stałym. Nie stanowi składnika gazy gazowej (nie jest rozpuszczony). Podczas reakcji nie zmienia się jego stężenie, tylko zmienia się masa – wielkość fazy stałej. Stężenie węgla w fazie stałej się nie zmienia (podobnie jak zawartość cukru w cukrze będzie taka sama, nawet jak kryształek urośnie lub zmniejszy się). Jeśli stężenie jakiejś fazy się nie zmienia, to jego wartość jest liczbą stałą. Możemy więc rozdzielić równanie stronami: stałe na jedną stronę, zmienne na drugą:

Ponieważ po lewej mamy teraz iloczyn stałych liczb, to wynik ich mnożenia także jest liczbą stałą. Oznaczmy tę nową stałą symbolem prim:

Ta nowa stała to właśnie używana w praktyce stała równowagi. Dotyczy wszystkich układów niejednorodnych, w których zachodzi odwracalna reakcja. Często słyszy się, że stężenie ciał stałych wynosi 1 i dlatego nie pojawiają się w równaniu stałej równowagi – nie jest to prawdą, ich stężenie jest uwzględnione w wartości stałej równowagi.
3) Stała równowagi a woda. To chyba najpospolitsze pytanie: czy wodę uwzględniamy w stałej równowagi? Odpowiedź na to pytanie będzie ściśle związane z poprzednim punktem. Rozpatrzmy dwie sytuacje, np. hydratację etenu w fazie gazowej w obecności katalizatora kwasowego:

Stała równowagi przyjmuje postać:

Ponieważ wszystkie składniki są w jednej fazie i stężenie wszystkich ulega zmianom, to wszystkie są po prawej stronie.
Druga sytuacja to hydroliza w roztworze wodnym octanu sodu. Załóżmy, że roztwór otrzymujemy przez rozpuszczenie 0,1 mol octanu sodu w 1 dm3 wody. W roztworze tym na jeden mol soli przypada ok. 550 moli wody. Na jeden jon pochodzący z dysocjacji tej soli przypada ok. 275 cząsteczek wody. Spójrzmy na równanie hydrolizy tej soli:

Jeśli hydrolizie ulegnie 1 na 10000 jonów octanowych, to przereaguje 1 na ok. 2 750 000 cząsteczek wody. Uznajemy więc, że stężenie wody w tym przypadku jest praktycznie stałe – praktycznie nie zmienia się, mimo tego, że bierze udział w reakcji. Zapiszmy równanie stałej równowagi hydrolizy:

Ponieważ jednak [H2O] jest liczbą stałą, to możemy wszystkie stałe przenieść na jedną stronę, a zmienne na drugą:

Stężenie wody ponownie jest uwzględnione w wartości stałej. Jednakże ma to swoje ograniczenia – tylko do sytuacji, w których można uznać stężenie wody za praktycznie niezmienne w trakcie zachodzenia reakcji, dlatego dotyczy to roztworów o niskich stężeniach – zazwyczaj poniżej 0,2 mol/dm3. Tak wyprowadzona stała nie dotyczy układów, w których woda nie jest jednocześnie środowiskiem reakcji, czyli nie stosuje się jej nawet do zbyt stężonych wodnych roztworów, bo nie można wtedy założyć niezmienności stężenia wody.
Podobnie gdyby woda była produktem jakiejś reakcji, ale prowadzonej w środowisku wodnym w rozcieńczonym roztworze, to także można uznać jej stężenie za niezmienne. Estryfikację prowadzi się w środowisku niewodnym – stężenie wody nie jest stałe, początkowo układ zazwyczaj w ogóle jej nie zawiera – dlatego nie można wrzucić jej stężenia do stałych, tylko jest po tej stornie równania, po której są zgromadzone zmienne.
Nie jest prawdą często powtarzana informacja, że wodę uwzględniamy tylko wtedy, gdy jest produktem! Patrz – pierwsza sytuacja.
12. Amoniak i kwas węglowy – znak mnożenia, czy dodawania?
To też problematyczny przypadek. Zapisy CO2+H2O i NH3+H2O są poprawne zawsze. Natomiast jeśli rozpatrujemy amoniak czy dwutlenek węgla w roztworze wodnym (kwas węglowy) – możemy zapisać wtedy NH3*H2O lub CO2*H2O. Z kolei w przypadku, gdy amoniak lub dwutlenek węgla opuszczają środowisko reakcji dopuszczalny jest tylko znak dodawania. Strzałka w górę oznaczająca wydzielanie się gazu nie jest obowiązkowa.
13. Katalizatory
Katalizator nie jest magicznym proszkiem – nie jest prawdą, że nie bierze udziału w reakcji. Skoro ją przyspiesza, to musi jakoś działać. Katalizatorami mogą być zarówno związki nieorganiczne, jak i organiczne, w tym skomplikowane substancje białkowe, czyli enzymy. W przypadku tych ostatnich często popełnianym błędem jest stwierdzenie, że enzym utlenia/redukuje/hydrolizuje itd. Nie – enzym uczestniczy w reakcji/przyspiesza/przeprowadza reakcję, ale skoro nie jest utleniaczem, ani reduktorem, to nie możemy powiedzieć, że utlenia itd – może katalizować/przyspieszać utlenianie/redukcję, ale nie redukuje i nie utlenienia, bo to terminy zarezerwowane dla utleniacza (np. O2) i reduktora. Podobnie niepoprawne jest stwierdzenie, że enzym hydrolizuje – to by oznaczało, że enzym ulega hydrolizie, czyli rozpada się na aminokwasy w reakcji z wodą. Enzym przeprowadza hydrolizę czegoś, ale nie hydrolizuje.
14. Wypieranie a strącanie
Nie należy mylić tych pojęć. Można wyprzeć metal z roztworu jego soli, wodór z kwasu – oznacza to otrzymanie wolnego metalu lub wodoru. Można wyprzeć kwas z soli – oznacza to otrzymanie wolnego kwasu. Można wyprzeć zasadę z soli – oznacza to otrzymanie wolnej zasady. Można strącić osad – często wyparcie jest związane z powstaniem związku o bardzo małej rozpuszczalności, czyli strącaniem. Ale należy pamiętać, że w przypadku reakcji powstawania osadów w reakcji z kwasami, np.:

Jest to reakcja strącania, ale nie jest to wyparcie. Czasem można spotkać stwierdzenie, że ta reakcja zachodzi, bo wodór wyparł mniej aktywną miedź – nie jest to prawda, nie otrzymaliśmy ani miedzi, ani substratem nie był wodór.
15. Izomeria optyczna
1) Enancjomery są swoimi odbiciami lustrzanymi – jeśli związek posiada więcej, niż jedno centrum chiralności (asymetryczny atom węgla), to jego izomer będący odbiciem lustrzanym ma przeciwne konfiguracje wszystkich centrów chiralności. Jeśli któreś centrum chiralności nie ma przeciwnych konfiguracji, to porównywane związki są wobec siebie diastereozimerami, a nie enancjomerami (odbiciami lustrzanymi). Enancjomery cukrów L i D mają więc przeciwne konfiguracje wszystkich centrów, a nie tylko ostatniego centrum. Jeśli w D-glukozie zmienić konfigurację tylko ostatniego centrum chiralności, to otrzymamy L-idoze, a nie L-glukozę.
2) W przypadku przypisywania form cyklicznych cukrów do szeregu D lub L należy spojdzeć na ostatnie centrum chiralności tak, by mieć najstarszy podstawnik (grupę OH) i najmłodszy (atom H) w linii prawa-lewa (strzałka). W przypadku poniżej mamy D-glukozę, bo najstarszy podstawnik jest po prawej stronie:

3) Związki nieczynne optycznie mają płaszczyznę symetrii – jeśli związek ma płaszczyznę symetrii, to ma równocenne grupy atomów. Nie jest prawdą, że płaszczyzna symetrii musi przechodzić pomiędzy atomami – może przechodzić przez atomy. Symetryczna jest zarówno cząsteczka etanu, jak i metanu czy propanu i acetonu – w tych ostatnich przecina ona atomy i dzieli cząsteczki na połówki, bo mamy nieparzystą liczbę atomów węgla w łańcuchu. Dzięki płaszczyźnie symetrii grupy CH3 w cząsteczka acetonu czy propanu są równocenne. Im więcej elementów symetrii ma cząsteczka (lub jon) związku, tym więcej równocennych grup ma. Wśród izomerów kwasu winowego płaszczyznę symetrii ma jedynie kwas mezo-winowy. Często popełnianym błędem jest stwierdzenie, że cząsteczki kwasu winowego mają płaszczyznę symetrii, dlatego może istnieć nieczynna optycznie forma mezo. Płaszczyznę symetrii ma włącznie forma mezo. To, że cząsteczkę można podzielić na połówki o identycznie połączonych atomach nie oznacza jeszcze, że istnieje płaszczyzna symetrii, bo w czynnych optycznie izomerach kwasu winowego jedna połówka cząsteczki nie jest lustrzanym odbiciem drugiej (nie ma przeciwnej konfiguracji) – dlatego są czynne optycznie. Przykładem nieczynnej optycznie formy mezo jest także ksylitol (C5H12O5), gdzie płaszczyzna symetrii przecina środkowy atom węgla.
Polecam materiały Rafała Szczypińskiego do poćwiczenia:
Określanie konfiguracji absolutnej centrów chiralności
Dozwolone przekształcenia wzorów izomerów optycznych narysowanych w projekcji Fischera
Przekształcanie wzorów stereoizomerów na projekcję Fischera
16. Amfoteryczność
Amfoteryczność dotyczy wielu substancji, ale trzeba pamiętać, że amfoteryczność dotyczy reakcji kwasowo-zasadowych, a związek jest amfoteryczny, jeśli jest zarówno kwasem, jak i zasadą i grupy kwasowe reagują z zasadami, zaś grupy zasadowe – z kwasami. Nie można udowadniać amfoteryczności reakcjami redoks, bo to inny typ reakcji. Amfoteryczność dotyczy związków chemicznych, a nie wolnych pierwiastków, bo wolne pierwiastki nie są kwasami, ani zasadami. Zasadą nie jest potas, tylko wodorotlenek potasu, kwasem nie jest siarka, tylko siarkowodór. Nie można mówić o amfoteryczności np. cynku, bo roztwarza sie w roztworach zasad i kwasu – to świadczy o jego właściwościach redukcyjnych, a nie amfoterycznych. Możemy mówić o amfoteryczności tlenku czy wodorotlenku cynku, a nie samego cynku jako metalu. Gdyby metale roztwarzały się w roztworach zasad ze względu na ich amfoteryczność, to także miedź by się musiała w nich roztwarzać, a się nie roztwarza.
Polecam: Tajemnica amfoteryczności
17. Wypieranie się mocnych kwasów
Stałe równowagi dysocjacji kwasowej, którymi dysponujecie, dotyczą jedynie rozcieńczonych wodnych roztworów. Nie można się nimi sugerować, mając do dyspozycji kwasy stężone i stałe sole, a nie wodne roztwory jednego i drugiego. Kwasy wypierają się wtedy, gdy w tych samych warunkach znacznie różnią się stopniem dysocjacji, np w wodnym roztworze kwas solny wypiera znacznie słabszy (słabo zdysocjowany) kwas octowy z octanu sodu czy potasu. Jeśli mamy do czynienia z kwasem mocnym i solą innego mocnego kwasu, to zwróćmy uwagę, że choć ich stałe dysocjacji znacznie się różnią – aż 10 000 razy w przypadku kwasu solnego i siarkowego, to jednak w rozcieńczonych roztworach nie wypierają się wzajemnie – bo oba są w podobnym stopniu zdysocjowane (bliskim 100%). Wyprą się wtedy, gdy w tych samych warunkach znacznie różnią się stopniem dysocjacji (mocą). Dlatego też NIE zachodzi wyparcie HNO3 z soli przez H2SO4 w wodnym środowisku, za to zachodzi, gdy potraktować stały KNO3 czy NaNO3 stężonym kwasem siarkowym(VI) – jest to jedna z metod otrzymywania HNO3. Jeśli stała dysocjacji kwasowej jest >20, to możemy założyć, że w rozcieńczonych roztworach wodnych kwas jest praktycznie całkiem zdysocjowany.
Polecam: O prawach wypieranie się kwasów, cz. II
oraz: film o wypieraniu się kwasów.
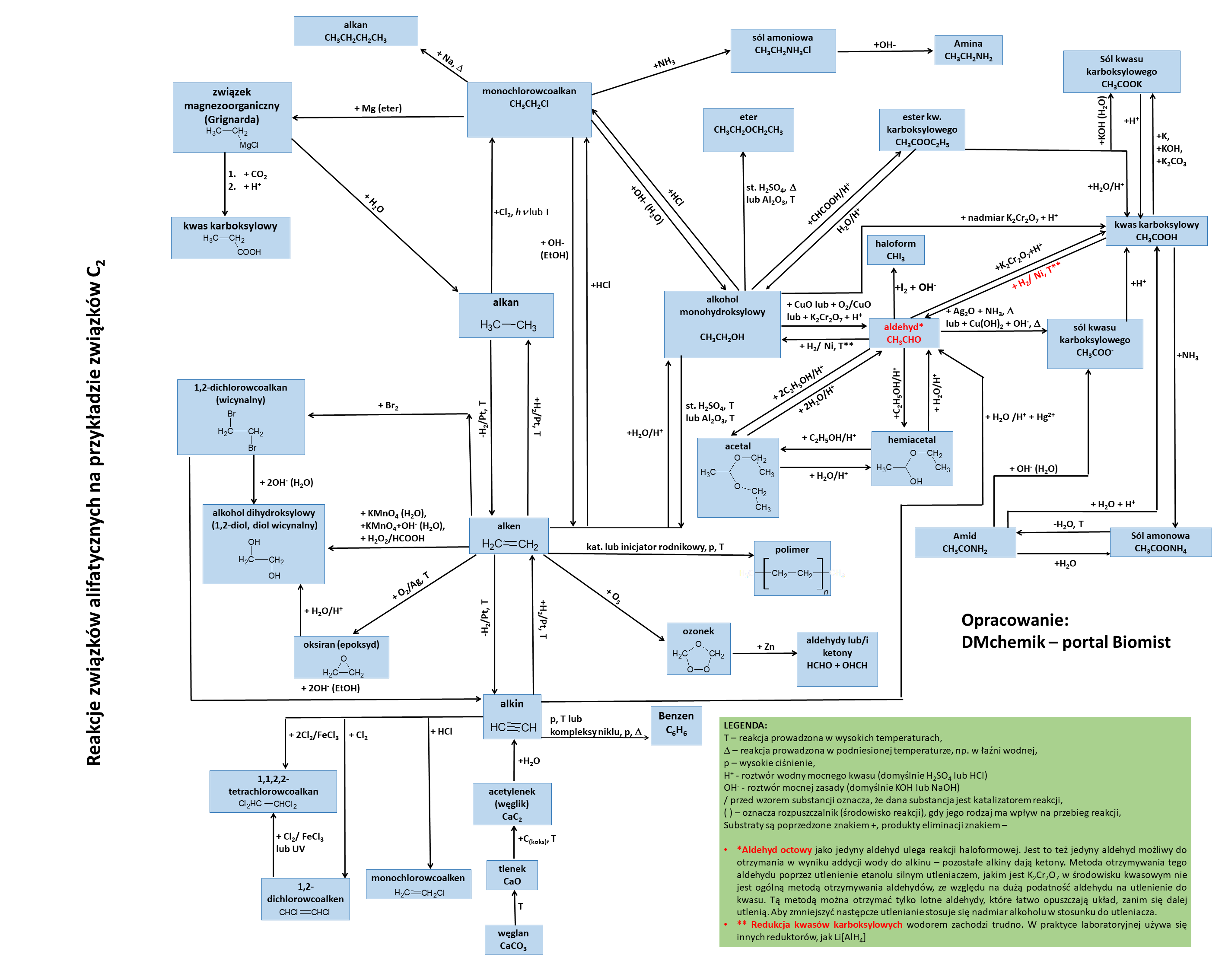
18. Kwas octowy i próba haloformowa (jodoformowa)
Próbie haloformowej/reakcji haloformowej ulegają metyloalkohole i metyloketony, które powstają przez utlenienie tych pierwszych. Aldehyd octowy jako jedyny z aldehydów ulega tej reakcji. Natomiast nie jest prawdą, że ulega jej kwas octowy, lub sole czy estry tego kwasu.
Dla zainteresowanych: próba haloformowa
Schemat reakcji związków organicznych alifatycznych: kliknij tutaj
{kind=link}
Zapraszam też na nasz kanał YT, gdzie można obejrzeć wiele doświadczeń 🙂
To tyle póki co ode mnie 🙂 Powodzenia 13.05! 🙂
ruchable artykulik dzieki !