Hemofilia nabyta (ang. acquired haemophilia – AH) jest ciężką skazą krwotoczną, spowodowaną nagłym wystąpieniem w osoczu chorego autoprzeciwciał skierowanych przeciwko czynnikowi krzepnięcia VIII. Występowanie tych autoprzeciwciał jest rzadkim zaburzeniem. Szacuje się, że pojawiają się one u 1 – 4 osób/ 1000000/ rok.
Hemofilia nabyta w przeciwieństwie do wrodzonej może rozwinąć się zarówno u mężczyzn, jak i u kobiet. Wyjątkiem jest populacja osób w wieku rozrodczym, w której omawiana skaza krwotoczna dotyka głównie kobiet.
Częstość występowania AH rośnie wraz z wiekiem. Najczęściej chorują osoby w 6 – 7 dekadzie życia. Występowanie AH u dzieci jest bardzo rzadkim zjawiskiem. Szacunkowa częstość występowania u osób poniżej 16 roku życia to 0,045/ 1000000/ rok.
Powyższe dane epidemiologiczne różnią się znacznie od tych charakteryzujących hemofilię wrodzoną, która dotyka głównie mężczyzn i może wywoływać krwawienia już we wczesnym dzieciństwie.
Skazę krwotoczną można podejrzewać u pacjenta, u którego doszło do spontanicznego krwawienia albo krwawienia powstałego po drobnym urazie czy zabiegu chirurgicznym. Jeżeli chory w swoim dotychczasowym życiu nie wykazywał tendencji do krwawień i wywiad rodzinny jest ujemny, należy rozważyć wystąpienie AH.
Etiologia hemofilii nabytej:
Etiologia hemofilii nabytej pozostaje niewyjaśniona. Szacuje się, że około połowa przypadków tej choroby rozwija się bez uchwytnej przyczyny. U pozostałej części chorych w chwili rozpoznania AH wykrywa się współistniejące choroby autoimmunologiczne lub nowotworowe. Szacuje się, że choroby autoimmunologiczne są przyczyną ok. 20% przypadków AH. W tej grupie chorób najczęściej wymienia się reumatoidalne zapalenie stawów oraz toczeń rumieniowaty układowy. Dość istotny odsetek (10%) pacjentów z AH stanowią osoby ze współistniejącą chorobą nowotworową, najczęściej są to rak płuca i gruczołu krokowego. Hemofilia nabyta może również rozwinąć się w ciąży i okresie poporodowym. Zaobserwowano również, że stosowanie niektórych leków (np. penicyliny) oraz szczepień sprzyja wystąpieniu omawianej skazy krwotocznej.
Objawy hemofilii nabytej:
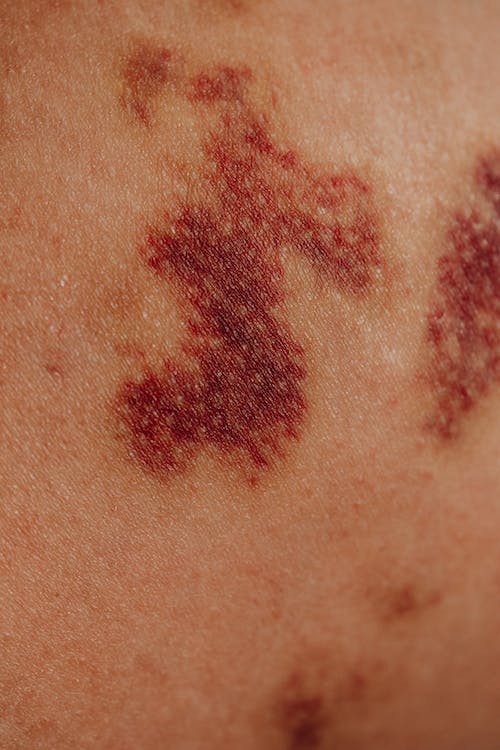
Obraz kliniczny hemofilii nabytej jest różnorodny pod względem ciężkości krwawień. Mogą to być niegroźne krwiaki, ale również ciężkie, zagrażające życiu krwawienia. AH objawia się ciężką skazą krwotoczną niemal u 90% pacjentów. Jednak krwawienia mogą być skutecznie kontrolowane, jeśli tylko choroba ta zostanie poprawnie i szybko zdiagnozowana oraz jeśli zostanie wdrożona odpowiednia terapia. Charakterystyczne w obrazie klinicznym AH są rozległe wynaczynienia krwi pod skórą i masywne krwawienia śluzówkowe.
Z przeglądu piśmiennictwa wynika, że wskaźnik śmiertelności w hemofilii nabytej jest wysoki. Jego wartość (przedstawiona jako wynik ilorazu liczby pacjentów chorych na AH, u których doszło do wczesnego zgonu i liczby wszystkich badanych pacjentów chorych na tę chorobę) zależnie od źródła waha się od 8 – 20%. Wartość tego wskaźnika wyraźnie wzrasta wraz z wiekiem pacjenta.
Diagnostyka laboratoryjna hemofilii nabytej:
Na początku diagnostyki laboratoryjnej AH należy wykonać podstawowe badania układu krzepnięcia, obejmujące ocenę hemostazy pierwotnej i wtórnej.
Ocena hemostazy pierwotnej:
Liczba płytek krwi (ang. platelet count – PLT) – aktualnie jest oznaczana przy użyciu cytometru z próbki krwi żylnej pobranej na antykoagulant. System zlicza cząsteczki o wielkości 2 – 37 µm3, które rozpraszają światło lasera. Jeżeli cząstki te są uprzednio wyznakowane barwnikami fluorescencyjnymi, to światło lasera powoduje ich wzbudzenie. Sygnał zostaje zarejestrowany przez detektory, ulega wzmocnieniu i zamieniany jest na sygnał cyfrowy [. Trombocytopenia, czyli obniżona liczba płytek krwi może wynikać ze zmniejszonego wytwarzania płytek w niektórych chorobach (np. niedokrwistość aplastyczna), z rozcieńczenia krwi (np. po masywnych krwotokach), z rozmieszczenia krwi (splenomegalia), z uszkodzenia układu immunologicznego (np. toczeń trzewny układowy) czy ze zniszczenia płytek (rozsiane wykrzepianie wewnątrznaczyniowe; DIC).
Norma: 150 – 400 G/l
Czas krwawienia (ang. bleeding time – BT) – badanie wykonywane jest poprzez niewielkie nacięcie skóry. Dokonuje się pomiaru czasu do zatrzymania krwawienia. BT zazwyczaj wydłużają trombocytopenia oraz strukturalne i/ lub czynnościowe choroby płytek krwi.
Ocena hemostazy wtórnej :
Czas protrombinowy (ang. prothrombin time – PT) wyrażony zwykle w formie międzynarodowego współczynnika znormalizowanego (ang. international normalized ratio – INR) – to próba czasu krzepnięcia zapoczątkowanego przez układ zewnątrzpochodny krzepnięcia. Test wykonuje się poprzez dodanie do osocza cytrynianowego komercyjnie dostępnej mieszaniny fosfolipidów i czynnika tkankowego oraz wapnia. Wydłużenie czasu PT może być spowodowane niedoborem czynników I, II, V, VII, X. Obecność inhibitorów tych czynników również może wydłużyć czas krzepnięcia. Wynik powinien być podawany jako wskaźnik INR .
Norma: 12 – 15s (PT)
Norma: 80 – 120% (INR)
Czas kaolinowo – kefalinowy, inaczej zzas częściowej tromboplastyny po aktywacji (ang. activated partial thrombin time – aPTT)– to próba czasu krzepnięcia zapoczątkowanego przez układ wewnątrzpochodny krzepnięcia. Test wykonuje się poprzez dodanie do osocza cytrynianowego kaolinu, co aktywuje cz. XII. Po aktywacji wewnątrzpochodnego układu krzepnięcia dodawane są fosfolipidy i wapń. Od tego momentu mierzy się czas, aż do wytworzenia skrzepu. Wydłużenie czasu aPTT może nastąpić w wyniku niedoborów czynników I, II, V, VIII, IX, X, XI, XII oraz w przypadku obecności inhibitorów tych czynników, antykoagulantu toczniowego albo heparyny.
Norma: 30 – 40s
Czas trombinowy (ang. thrombin time – TT) – test ten wykonuje się poprzez dodanie do osocza trombiny, co bezpośrednio wywołuje utworzenie skrzepu. Czas TT wydłuża się w przypadku obecności heparyny, lub z powodu jakościowego czy ilościowego niedoboru fibrynogenu.
Norma: ok.15 s
Najbardziej typowym wynikiem przesiewowych badań laboratoryjnych dla hemofilii nabytej jest wyizolowane przedłużenie czasu aPTT (zazwyczaj 2 – 3– krotnie) przy prawidłowym czasie PT i TT oraz prawidłowej liczbie płytek krwi. Jednak należy wziąć pod uwagę fakt, że podobne wyniki można otrzymać w przypadku obecności w osoczu inhibitora skierowanego przeciwko innym niż cz. VIII czynnikom krzepnięcia wewnątrzpochodnej drogi krzepnięcia.
Hemofilia nabyta nierzadko jest mylona z innymi skazami krwotocznymi, między innymi z zespołem rozsianego wykrzepiania wewnątrznaczyniowego (ang. Disseminated intravascular coagulation – DIC). Jednak szczegółowa analiza laboratoryjna umożliwia zdiagnozowanie DIC na podstawie nie tylko przedłużonego czasu aPTT, ale również wydłużenia czasu PT, trombocytopenii (małopłytkowości), a także obniżonego stężenia fibrynogenu i wzrostu stężenia produktów degradacji fibryny. Rozpoznanie hemofilii nabytej powinno również uwzględniać wykluczenie obecności zaburzeń, takich jak występowanie antykoagulantu tocznia, choroby von Willebranda, dysfibrynogenemii, hemofilii wrodzonej, niespecyficznych inhibitorów innych niż cz. VIII oraz podanie heparyny.
Do błędów w diagnostyce tej choroby przyczynia się fakt, że nie u wszystkich pacjentów cierpiących na AH można zaobserwować znaczne wydłużenie czasu aPTT czy ciągłych krwawień. Dlatego, aby zwiększyć prawdopodobieństwo zdiagnozowania AH bardzo ważna jest współpraca pomiędzy klinicystami, diagnostami laboratoryjnymi i specjalistycznymi laboratoriami hematologicznymi doświadczonymi w oznaczaniu miana inhibitora.
Test korekcji aPTT
Skoro przedłużony czas aPTT można przypisać zarówno niedoborom czynników krzepnięcia, obecności antykoagulantu toczniowego, jak i leczeniu heparyną, konieczne jest przeprowadzenie dalszych badań mających na celu wykrycie przyczyny wydłużenia czasu kaolinowo – kefalinowego.
Zwyczajowo, w celu odróżnienia obecności inhibitorów od niedoborów czynników krzepnięcia stosuje się test korekcji aPTT. W teście korekcji mierzy się czas aPTT mieszaniny równych objętości osocza badanego i osocza prawidłowego po inkubacji w 37°C. W przypadku podejrzenia nabytych inhibitorów cz. VIII inkubacja powinna trwać co najmniej 1 godzinę. Na inhibitory wpływa zarówno czas, jak i temperatura wykonywania testu. Dlatego słabe autoprzeciwciała anty – VIII mogą wymagać do 2 godzin inkubacji, przy czym silnym inhibitorom wystarczają zaledwie minuty.
Jeżeli w teście korekcji nie dojdzie do wyrównania przedłużonego czasu aPTT mamy do czynienia z autoprzeciwciałami przeciwko cz. VIII. Jeżeli natomiast po inkubacji zaobserwujemy wyrównanie przedłużonego czasu aPTT, to możemy mówić o niedoborze któregoś spośród czynników krzepnięcia.
Należy pamiętać o tym, że od każdej reguły występują wyjątki i dlatego korekcja wydłużonego czasu kaolinowo – kefalinowego nie wyklucza AH typu A. Jeżeli obraz kliniczny sugeruje nabytą, ciężką skazę krwotoczną to należałoby przeprowadzić badania, zarówno w kierunku wykrycia obecności inhibitorów, jak i w kierunku innych potencjalnych przyczyn objawów skazy krwotocznej. Niezależnie od wyników testu korekcji aPTT wymagane jest przeprowadzenie dalszych badań. Równoległe oznaczenie aktywności czynników krzepnięcia znacznie ułatwia postawienie wczesnej diagnozy.
Wyeliminowanie obecności heparyny w osoczu badanym, jako przyczyny przedłużonego czasu aPTT
Kolejnym etapem badań laboratoryjnych, po wykonaniu testu korekcji aPTT, którego wynik sugeruje obecność inhibitorów w osoczu, jest sprawdzenie czy próbka nie była zanieczyszczona heparyną. W tym celu należy wykonać pomiar czasu protrombinowego i reptylazowego. Jeśli heparyna jest obecna w osoczu, to czas protrombinowy będzie wydłużony. Natomiast czas reptylazowy pozostanie w normie, gdyż wykrzepanie fibrynogenu przez reptylazę nie jest hamowane przez heparynę.
Wyeliminowanie obecności antykoagulantu toczniowego jako przyczyny przedłużonego czasu aPTT w osoczu badanym
Antykoagulant toczniowy również może być przyczyną wydłużenia aPTT, który nie ulega korekcji w teście z prawidłowym osoczem. Obecność tego antykoagulantu skutkuje też obniżeniem stężenia wewnątrzpochodnych czynników krzepnięcia i pozytywnym wynikiem testu Bethesda.
Antykoagulant toczniowy nie zależy od temperatury. Powinny być podejmowane specyficzne testy do jego wykrywania, co ugruntowałoby diagnostykę różnicową.
W bardziej złożonych przypadkach klinicznych, w jednej probówce mogą występować autoprzeciwciała przeciwko cz. VIII i antykoagulant toczniowy. W takiej sytuacji przydatny jest test ELISA wykrywający inhibitory cz. VIII, który pozwoli ostatecznie zdecydować pomiędzy obecnością antykoagulantu tocznia w osoczu pacjenta i nabytymi przeciwciałami anty – VIII.
Pomiar czynników krzepnięcia
U pacjentów z objawami klinicznymi sugerującymi AH oraz przedłużonym czasem aPTT dokonuje się pomiaru czynników krzepnięcia: VIII, IX, XI, XII. Wyizolowane, niskie stężenia cz. VIII kieruje diagnostykę w stronę AH.
W niektórych przypadkach, wszystkie wyżej wymienione czynniki krzepnięcia wykazują zmniejszone stężenie. Jest to obrazem artefaktu powstałego in vitro w probówce z osoczem w wyniku wyczerpania cz. VIII przez inhibitory.
Antykoagulant toczniowy również może wpływać na obniżenie stężenia czynników krzepnięcia poprzez hamowanie fosfolipidów w teście. W takich okolicznościach pomiar czynników krzepnięcia powinien być powtórzony w serii rozcieńczeń osocza, co osłabi efekt działania inhibitora albo antykoagulantu toczniowego na stężenie poszczególnych czynników.
Określenie miana inhibitora
Ilość inhibitora w osoczu ocenia się na podstawie stopnia inaktywacji cz. VIII w mieszaninie inkubacyjnej osocza chorego ze źródłem cz. VIII. Do oznaczania miana przeciwciał stosuje się metodę Bethesda. Polega ona na inkubacji równych objętości osocza badanego z osoczem prawidłowym (będącym źródłem cz. VIII o stężeniu równym 100 jednostek cz. VIII/ dl) przez 2 godz. w temp. 37°C.
1 jednostka Bethesdy (1BU) – taka ilość inhibitora w osoczu, która po inkubacji inaktywuje 50% cz. VIII w osoczu zdrowym.
Miano inhibitora określa się jako niskie, jeżeli wynosi mniej niż 10 BU. 10 – 20 BU oznacza średnie miano tych przeciwciał. Natomiast wysokie miano inhibitora występuje, gdy wynik testu będzie wyższy niż 20 BU.
Test Bethesda został stworzony do oceny miana alloprzeciwciał cz. VIII, wykazujących kinetykę typu 1. Przebieg inaktywacji cz. VIII jest bezpośrednio zależny od stężenia inhibitora. Natomiast autoprzeciwciała cz. VIII nie wykazują liniowej zależności pomiędzy inaktywacją cz. VIII a stężeniem inhibitora. Z tego powodu test Bethesda nie umożliwia oszacowania prawdziwej siły hamowania nabytych inhibitorów cz. VIII.
Sporadycznie, przy obniżonej aktywności cz. VIII, inhibitor nie ujawnia się od razu po zakończeniu testu, natomiast może zostać wykryty po kilku dniach inkubacji. W takiej sytuacji należy przeprowadzić ponowne badania w celu potwierdzenia obecności inhibitora. Miano wieprzowego inhibitora cz. VIII powinno być określone, w przypadku gdy taka opcja terapeutyczna jest możliwa. Należy pamiętać, że zazwyczaj miano wieprzowych przeciwciał przeciwko cz. VIII jest niższe niż ludzkich.
Patogeneza hemofilii nabytej:
Hemofilia nabyta jest chorobą autoimmunologiczną wywołaną przez przeciwciała upośledzające funkcje czynnika VIII. Przeciwciała te określa się jako krążące antykoagulanty albo inhibitory czynnika VIII.
W literaturze opisano też niezwykle rzadkie przypadki hemofilii nabytej spowodowanej wytworzeniem przeciwciał skierowanych na inne czynniki krzepnięcia: cz. IX, cz. II, cz. V, cz. X, cz. XI, cz. XIII.
Czynnik VIII jest β2 – globuliną o masie cząsteczkowej 265 kDa, kodowaną przez gen znajdujący się na długim ramieniu chromosomu X (Xq28). Składa się on z 6 podjednostek (A1, A2, B, A3, C1, C2) tworzących dwa łańcuchy: lekki i ciężki. Synteza cz. VIII odbywa się głównie w wątrobie, w mniejszym stopniu w komórkach śledziony i węzłów chłonnych. 95% cząsteczek cz. VIII jest niekowalencyjnie związanych z cz. von Willebranda i krąży w osoczu w postaci kompleksu cz. VIII – cz. vWF. Czynnik von Willebranda chroni cz. VIII przed aktywacją za pomocą cz. Xa oraz przed inaktywacją przez białko C. Czynnik VIII w kompleksie z cz. vWF ma kilkakrotnie dłuższy okres półtrwania niż krążący samodzielnie (9 – 18 godzin).
Czynnik VIII odgrywa istotną rolę w procesie krzepnięcia krwi. Spadek aktywności i stężenia cz. VIII, wywołany obecnością przeciwciał skierowanych przeciwko cz. VIII, jest poważnym zagrożeniem dla zdrowia i życia pacjenta.
Skutkiem występowania autoprzeciwciał anty – VIII są zaburzenia krzepnięcia krwi. Dokładny mechanizm działania tych przeciwciał zależy od tego, z którą podjednostką cz. VIII się zwiążą. W patogenezie hemofilii nabytej znaczenie mają przeciwciała anty – A2, anty – A3 oraz przeciwciała anty – C2. W przypadku, gdy autoprzeciwciała wiążą się z podjednostką A2 w łańcuchu ciężkim lub A3 w łańcuchu lekkim cz. VIII, dochodzi do zaburzenia tworzenia tenazy. Natomiast przeciwciała anty – C2 powodują upośledzenie oddziaływań cz. VIII z fosfolipidami oraz prawdopodobnie blokują wiązanie cz. VIII z cz. vWF. Struktura cz. VIII ma odzwierciedlenie w swojej funkcjonalności. W skład łańcucha ciężkiego wchodzą podjednostki: A1, A2 i B, natomiast łańcucha lekkiego: A3, C1 i C2. Podjednostki A2 i A3 posiadają miejsca wiążące aktywny cz. IX, natomiast C2 zajmuje się wiązaniem cz. vWF i przytwierdzeniem błony fosfolipidów do cząsteczki cz. VIII. Miejscami aktywnymi na czynniku VIII, z którym wiążą się autoprzeciwciała są aminokwasy 454 – 509 i 593 na podjednostce A2, 1804 – 1819 na A3 oraz 2181 – 2243 na C2 .
Aktualny stan wiedzy potwierdza, że zarówno u chorych na hemofilię wrodzoną po podaniu dożylnym cz. VIII, jak i hemofilię nabytą przyczyną skłonności do krwawień jest obecność inhibitorów cz. VIII. Inhibitory te wykazują w obu jednostkach chorobowych pewne podobieństwa i różnice.
Cechą wspólną obu typów przeciwciał jest ich poliklonalny charakter oraz przynależność do klasy IgG immunoglobulin. Zarówno alloprzeciwciała jak i autoprzeciwciała nie wiążą dopełniacza i nie powodują reakcji alergicznych.
Główna różnica pomiędzy inhibitorami cz. VIII w hemofilii nabytej i wrodzonej leży w metodzie ich działania. Polega ona na tym, że autoprzeciwciała są skierowane przeciwko pojedynczym epitopom na cząsteczce cz. VIII. Są to epitopy znajdujące się na podjednostce A2, A3 lub częściej na podjednostce C2. Natomiast alloprzeciwciała zazwyczaj są skierowane przeciwko obu podjednostkom jednocześnie, zarówno A2 jak i C2 (czasami również przeciwko podjednostce A3).
Kinetyka oddziaływań cz. VIII z przeciwciałami charakterystycznymi dla hemofilii nabytej jest odmienna od obserwowanej w hemofilii wrodzonej powikłanej występowaniem alloprzeciwciał. Przeciwciała występujące u pacjentów z hemofilią wrodzoną zazwyczaj całkowicie inaktywują cz. VIII (kinetyka typu I). Przebieg inaktywacji pozostaje w bezpośredniej zależności od stężenia inhibitora. Natomiast w nabytej hemofilii, nawet przy bardzo dużym mianie autoprzeciwciał, w osoczu chorego wykrywa się resztkową aktywność cz. VIII (kinetyka typu II). W tym przypadku nie jest możliwe stwierdzenie liniowej zależności pomiędzy stopniem inaktywacji cz. VIII a stężeniem inhibitora.
Z tego powodu test Bethesda wykorzystywany do pomiaru in vitro stężenia inhibitora cz. VIII, może zaniżać aktualną siłę hamowania in vivo. Co za tym idzie, znajomość kinetyki działania autoprzeciwciał może mieć praktyczne znaczenie w monitorowaniu i leczeniu hemofilii nabytej.
Co ciekawe inhibitory cz. VIII zostały wykryte u ok. 15% zdrowych osób, które nie chorowały ani na hemofilię nabytą, ani na żadną powiązaną z nią chorobę czy koagulopatię. Autoprzeciwciała u tych osób, przeważnie klasy IgG1 i IgG4, wiążą się zazwyczaj z domeną C1 cz. VIII.
Oznacza to, że nie wszystkie przeciwciała skierowane przeciwko cz. VIII są patogenne i znacząco zmniejszają jego aktywność.
Z najnowszych danych wynika, że jedną z przyczyn występowania przeciwciał skierowanych na cz. VIII jest obecność polimorfizmu genu kodującego antygen CTLA – 4 (ang. cytotoxic T lymphocyte antigen – 4). Antygen CTLA – 4 to białko transbłonowe obecne na limfocytach T. Białko to odgrywa kluczową rolę w negatywnej regulacji odpowiedzi immunologicznej indukowanej obecnością egzogennego antygenu.
Badania pokazują, iż CTLA – 4 nie jest stale obecne na powierzchni komórek T. Transport tej immunoglobulinopodobnej cząsteczki z cytoplazmy na powierzchnię limfocytów T zachodzi jedynie po aktywacji tych komórek. Prawidłowa aktywacja limfocytów T opiera się na dwóch niezależnych sygnałach przekazywanych przez komórki prezentujące antygen (ang. antigen presenting cells – APC) – dostarczanych przez receptor TCR (ang. T – cell receptor) (I sygnał) i cząstki kostymulujące (II sygnał). Drugi sygnał kostymulujący może stanowić kompleks antygenu CTLA – 4 z glikoproteinowymi molekułami CD28 i B7 . Połączenie się CTLA – 4 z B7 występującym na błonie komórkowej monocytów, makrofagów, komórek dendrytycznych, tymocytów, limfocytów T i B prowadzi do zahamowania syntezy IL – 2 przez limfocyty T oraz do ograniczenia proliferacji i różnicowania się tych komórek.
Dotychczasowe doświadczenia potwierdzają, że istnieje związek pomiędzy hemofilią nabytą a obecnością polimorfizmu genu kodującego antygen CTLA – 4. Gen CTLA – 4 jest zlokalizowany na chromosomie 2 (2q33).
Dotychczas poznano trzy warianty polimorficzne tego genu:
-w promotorze w pozycji 318 od kodonu start ATG
– w eksonie 1 w pozycji 49 skutkującym substytucją Ala – 17Thr
-w eksonie 3 w regionie niepodlegającym translacji
Wystąpienie określonych wariantów polimorficznych genu CTLA – 4 powoduje spadek ekspresji kodowanego przez ten gen białka i/ lub zaburzenia jego funkcji, co skutkuje wzmożoną aktywnością limfocytów T (rozwojem autoimmunizacji). U chorych na AH zaobserwowano wzrost częstości występowania niektórych alleli HLA klasy II w porównaniu z grupą osób zdrowych.
Antygeny głównego układu zgodności tkankowej (ang. major histocompatibility complex – MHC), u człowieka nazywane HLA, kodowane są przez geny zlokalizowane na krótkim ramieniu chromosomu 6. Aktualnie MHC stanowi jeden z najintensywniej badanych regionów ludzkiego genomu. Główny układ zgodności tkankowej składa się z więcej niż 200 genów. W ich obrębie można wyróżnić 3 podregiony. Produkty wspomnianych genów klasyfikowane są odpowiednio jako antygeny klasy I (HLA – A – B i – C), klasy II (HLA – DR, – DQ i – DP) oraz klasy III. Cząsteczki klasy I można znaleźć na powierzchni komórek większości tkanek. Z kolei charakterystyczną cechą MHC klasy II jest obecność na komórkach biorących udział w odpowiedzi immunologicznej – komórkach APC. Produkty genów układu HLA pełnią istotną rolę w prezentacji antygenowej limfocytom T, kontrolują również niektóre reakcje wrodzonego układu odpornościowego. Pomimo licznych doświadczeń przeprowadzonych w celu określenia wpływu MHC na układ odpornościowy, nadal pozostaje do wyjaśnienia kwestia mechanizmu wiążącego antygeny głównego układu zgodności tkankowej z chorobami autoimmunologicznymi. Pewnym jest jedynie, że MHC wywiera wpływ na autoimmunizację poprzez regulowanie funkcji komórek APC. Odnotowano fakt, że specyficzne allele MHC klasy II determinują kierunek poszczególnym autoantygenom, co skutkuje wystąpieniem określonej choroby. Dostępne w literaturze wyniki badań nad powiązaniem polimorfizmu HLA z chorobami autoimmunologicznymi (np. cukrzyca typu I) pozwalają wysnuć wniosek, że układ HLA ma predyspozycje zarówno do rozwoju chorób autoimmunologicznych, jak i do ochrony przed nimi.
Wykazano, że wzrost wrażliwości na inhibitory rozwijające się w ciężkiej wrodzonej hemofilii A jest powiązany z allelami HLA DRB1*1501, DQA1*0102, DQB1*0602.
W celu porównania hemofilii wrodzonej z nabytą Pavlova i wsp. [Pavlova A., Zeitler H., Scharrer I., Brackmann H.- H., Oldenburg J.: HLA genotype in patients with acquired haemophilia A. Haemophilia, 2010, 16: 107- 112] przeprowadzili badania nad związkiem AH z HLA klasy I (HLA – A, HLA – B, HLA – C) oraz klasy II (HLA – DRB1 i HLA – DQB1). Badania przeprowadzono na 57 chorych na AH. U żadnego z nich nie zanotowano związku pomiędzy obecnością alleli klasy I a wystąpieniem AH. Natomiast analiza alleli klasy II ukazała istotny statystycznie wzrost częstości występowania alleli DRB1*16 i DQB1*0502 u badanych pacjentów w porównaniu z grupą zdrowych Europejczyków. Zaobserwowano również, że allele DRB1*15 i DQB1*0602 występują z mniejszą częstością u chorych na AH niż u zdrowych.
Autorzy cytowanej pracy porównali otrzymane wyniki z badaniami Oldenburga i wsp. [Oldenburg J., Picard J. K., Schwaab R., Brackmann H. H., Tuddenham E. G. D., Simpson E.: HLA genotype of patients with severe haemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost, 1997, 77: 238- 242] przeprowadzonymi u chorych na hemofilię wrodzoną. Pavlova i wsp. [Pavlova A., Zeitler H., Scharrer I., Brackmann H.- H., Oldenburg J.: HLA genotype in patients with acquired haemophilia A. Haemophilia, 2010, 16: 107- 112]wysnuli śmiałą hipotezę, iż allele DRB1*16 i DQB1*0502 będące wskaźnikami wysokiego ryzyka w AH są allelami niskiego ryzyka hemofilii wrodzonej. Analogicznie DRB1*15 i DQB1*0602 występujące z mniejszą częstością w AH są allelami wysokiego ryzyka hemofilii wrodzonej. Dokładny patomechanizm tych zależności do tej pory nie został poznany. Mimo to, mechanizm zaproponowany przez Pavlova i wsp. [Pavlova A., Zeitler H., Scharrer I., Brackmann H.- H., Oldenburg J.: HLA genotype in patients with acquired haemophilia A. Haemophilia, 2010, 16: 107- 112] jest obecnie uznawany za mechanizm powstawania inhibitorów cz. VIII u chorych na AH i wrodzoną.
Być może badania nad podłożem genetycznym oraz immunologicznym AH pozwolą w przyszłości na lepsze zrozumienie patomechanizmu uruchomionego w organizmie chorych na tę skazę krwotoczną i umożliwią wdrożenie skuteczniejszego leczenia . Wielką nadzieję pokłada się w badaniach nad związkiem między obecnością alleli HLA oraz wariantów polimorficznych antygenu CTLA – 4 a klinicznymi i immunologicznymi cechami AH. Zależności te nie zostały jeszcze potwierdzone w badaniach klinicznych.
Leczenie hemofilii nabytej:
Wybór metody leczenia hemofilii nabytej zależy przede wszystkim od obrazu klinicznego oraz miana inhibitora we krwi chorego. Główne cele terapii tej skazy krwotocznej to leczenie i profilaktyka krwawień (terapia hemostatyczna), eliminacja inhibitorów cz. VIII (terapia immunosupresyjna) oraz leczenie chorób współistniejących. U chorych na ciężką postać AH krwawienia zagrażające życiu mogą być skutkiem nawet banalnych zabiegów, takich jak zastrzyki domięśniowe czy pobieranie krwi tętniczej. Dlatego po zdiagnozowaniu u pacjenta omawianej choroby należy unikać inwazyjnych procedur w przebiegu leczenia. Ważnym jest także, by nie podawać choremu leków wpływających na funkcje płytek krwi (np. aspiryny).
Nie wszyscy pacjenci z diagnozą hemofilii nabytej wymagają terapii hemostatycznej. Jednak należy pamiętać, że ryzyko ciężkich krwawień utrzymuje się tak długo jak długo we krwi znajdują się inhibitory.
Jak wynika z obserwacji klinicznych, hemofilia nabyta jest dość heterogenną jednostką chorobową. Co za tym idzie optymalne leczenie dla jednego chorego na AH może nie przynieść pożądanych skutków u drugiego pacjenta.
Autorzy: mgr Katarzyna Chojnacka, diagnosta laboratoryjny oraz mgr Aleksadnra Maguza, diagnosta laboratoryjny
Piśmiennictwo:
1. Collins PW, Macartney N, Davies R, et al. A population based, unselected, consecutive kohort of patients with acquired haemophilia. Brit J Haematol. 2004;124:86-90.
2. Collins PW, Hirsh S, Baglin T, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors Organisation. Blood. 2007;109:1870-1877.
3. Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost. 1981;45:200-203.
4. Kessler C, Nemes L. Acquired inhibitors to factor VIII. In: Inhibitors in patients with haemophilia. Ed. Rodriguez-Merchan E.C. I Lee C.A. Blackwell Science Oxford 2002:98-114.
5. Windyga J. Acquired haemophilia. J Transf Med. 2010;4:131-132.
6. Collins PW, Percy CL. Advances in the understanding of acquired haemophilia A: implications for clinical practice. Brit J Haematol. 2010;148:183-194.
7. Windyga J, Chojnowski K, Klukowska A, Łętowska M, Mital A, Musiał J, Podolak-Dawidziak M, Undas A, Zdziarska J, Zawilska K, w imieniu Grupy Roboczej ds. Hemostazy Polskiego Towarzystwa Hematologów i Transfuzjologów. Polskie zalecenia postępowania w nabytej hemofilii A. Medycyna Praktyczna. 2011;10:1-8.
8. Huth-Kühne A, Baudo F, Collins P, Ingerslev J, Kessler CM, Levesque H, Castellano MEM, Sima M, ST-Louis J. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica. 2009;94:566-575.
9. Nemes L, Pitlik E. Ten years experience with immune tolerance induction therapy in acquired hemophilia. Haematologica. 2003;88(suppl. 12):64-68
10. Baudo F., Caimi T., De Catalado F.: Diagnosis and treatment of acquires haemophilia. Haemophilia, 2010, 16: 102-106.
11. Tufano A., Coppola A., Guida A., Cimino E., De Gregorio A. M., Cerbone A. M., Di Minno G.: Acquired haemophilia a in the elderly: case reports. Curr Gerontol Geriatr Res., 2010, 2010: 927503.
12 Pavlova A., Zeitler H., Scharrer I., Brackmann H.-H., Oldenburg J.: HLA genotype in patients with acquired haemophilia A. Haemophilia, 2010, 16: 107- 112.
13. Oldenburg J., Picard J. K., Schwaab R., Brackmann H.H., Tuddenham E. G. D., Simpson E.: HLA genotype of patients with severe haemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost, 1997, 77: 238-242.
14. Pavlova A., Diaz- Lacava A., Zeitler H., Satoguina J., Niemann B., Krause M., Scharrer I., Hoerauf A., Wienker T., Oldenburd J.: Increased frequency of the CTLA-4 49 A/G plymorphism in patients with acquired haemophilia A compared to healthy controls. Haemophilia, 2008, 14: 335-360.
15. Porwit A., McCullough J., Erber W. N.: Blood and bone marrow pathology, wyd. 2, Elsevier Churchill Livingstone, 2011, 479- 483.
16. Damjanov I.: Patofizjologia, wyd. 1, pod red.: Bręborowicz A., Thor P. J., Winnicka M. M., Wrocław, Elsevier Urban&Partner, 2010, 213- 214.
17. Bouvry P., Recloux P.: Acquired hemophilia. Haematologica, 1994, 79: 550- 556.
18. Ma A. D., Carrizosa D.: Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program, 2006: 432- 437.
19. Collins P. W.: Management of acquired haemophilia A- more question than answers. Blood Coagul Fibrinolysis, 2003, 14(1): 23- 27, Ma A. D., Carrizosa D.: Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program, 2006: 432- 437.
20. Lossing T. S., Kasper C. K., Feinstein D. I.: Detection of factor VIII inhibitors with the partial thromboplastin time. Blood, 1977, 49: 793- 797.
21. Kazmi M. A., Pickering W., Smith M. P., Holland L. J., Savidge G. F.: Acquired haemophilia A: errors in the diagnosis. Blood Coagul Fibrinolysis, 1998, 9: 623- 628.
22. Greaves M., Cohen H., MacHin S. J., Mackie I.: Guidelines on the investigation and management of the antiphospholipid syndrome. Br J Haematol, 2000, 109: 704- 715.
23. Sahud M. A., Pratt K. P., Zhukov O., Qu K., Thompson A. R.: ELISA system for detection of immune responses to FVIII: a study of 246 samples and correlation with the Bethesda assay. Haemophilia, 2007, 13: 317- 322.
24. Astermark J., Voorberg J., Lenk H., Dimichele D., Shapiro A., Tjonnfjord G., Berntorp E.: Impact of inhibitor epitope profile on the neutralizing effect against plasma- derived and recombinant factor VIII concentrates in vitro. Haemophilia, 2003, 9: 567- 572.
25. Kasper C. K.: Laboratory tests for factor VIII inhibitors, their variation, significance and interpretation. Blood Coagul Fibrinolysis, 1991, 2(1): 7- 10.
26. Czyż A, Kopydłowska A.: Nabyta hemofilia A- diagnostyka i leczenie. Przew Lek, 2003, 6: 101-105.
27. Bouvry P., Recloux P.: Acquired hemophilia. Haematologica, 1994, 79: 550- 556.
28. Gawryl M. S., Hoyer L. W.: Inactivation of factor VIII coagulant activity by two different types of human antibodies. Blood, 1982, 60: 1103- 1109.
29. Verbruggen B., Novakova I., Wessels H., Boezeman J., van der Berg M., Mauser- Bunschoten E. : The Nijmegen modification of the Bethesda assay for factor VIII:C inhibitors: improved specificity and reliability. Thromb Haemost, 1995, 73: 247- 251.
30. Morrison A. E., Ludlam C. A., Kessler C.: Use of porcine factor VIII in the treatment of patients with acquired hemophilia. Blood, 1993, 81: 1513- 1520.
31. Delgado J., Jimenez-Yuste V., Hernandez-Navarro F., Villar A.: Acquired haemophilia: review and meta- analysis focused on therapy and prognostic factors. Br J Haematol, 2003, 121: 21–35.
32. Johansen R. F., Sorensen B., Ingerslev J.: Acquired haemophilia: dynamic whole blood coagulation utilized to guide haemostatic therapy. Haemophilia, 2006, 12: 190–197.
Co to jest Eiopatogeneza I Diagnostyka Laboratoryjna Hemofilii Nabytej? – Ten artykuł to odpowiedź na te pytanie które mnie nurtowało. Polecam!!!